Remission of secondary membranous nephropathy in a patient with Kimura disease after surgical resection
Article information
Abstract
Kimura disease (KD) is an eosinophilic, granulomatous, benign, chronic inflammatory disease with an unknown etiology. A 33-year-old woman visited our hospital because of a palpable, left subclavian mass, a left scapulo-anterior pseudoaneurysm, and nephrotic syndrome. Her subclavian lymph node biopsy examination result was consistent with KD, and results of a renal biopsy indicated secondary membranous nephropathy. After renal histological examination confirmed nephropathy, treatment with prednisolone and cyclosporine was initiated, which was maintained for over 1 year. However, this therapy only provided a transient improvement in proteinuria. One year after commencing the treatment, both proteinuria and azotemia aggravated as the left axillary mass doubled in size. Finally, the mass was surgically excised, following which the azotemia rapidly normalized and proteinuria resolved within 1 month. This case shows that tumor resection in a patient with KD with secondary nephropathy may resolve secondary renal manifestations. Furthermore, reversible renal dysfunction may be caused by unknown secreted molecules.
Introduction
Kimura disease (KD) presents as a subcutaneous mass, most frequently occurring in the head and neck regions. The disease is characterized by a benign, angiolymphomatous proliferation with eosinophilic infiltration. The histological characteristics of the disease are hyperplastic lymphoid tissue that contains well-developed lymphoid follicles, marked infiltration of eosinophils, vascular proliferation, and fibrosis. It is also associated with the development of renal disease, especially nephrotic syndrome [1]. Previously, surgical resection was the treatment of choice. However, treatments with steroid and immunosuppressive agents were also reported to produce beneficial effects [2], although discontinuation of steroid often results in recurrence of the masses [3]. In patients with renal manifestations, however, surgical resection is usually not the first-choice treatment. We herein report the case of secondary membranous nephropathy (MN) associated with KD which recovered after surgical resection of the mass.
Case report
A 33-year-old woman presented with a soft-tissue mass in her left axillary area. A percutaneous biopsy was performed, and histological findings showed atypical lymphoid hyperplasia with many eosinophilic infiltrations, which are consistent with KD (Fig. 1). Neither proteinuria nor hypereosinophilia was noted. One-month treatment with oral prednisolone could not diminish size of the mass. One year after the diagnosis of KD, a 6-cm, left scapulo-anterior pseudoaneurysm was noted (Fig. 1), which required a stent-graft insertion. Following the graft insertion, the size of pseudoaneurysm decreased. Hypereosinophilia (12,450/mm3) was noted during this period. Treatment with prednisolone was continued for 1 year, and eosinophilia gradually improved to<500/mm3 within this period.
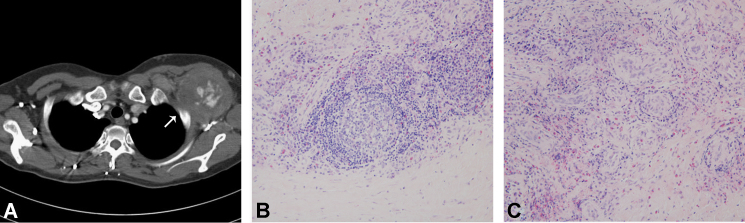
Left subclavian mass consistent with Kimura disease. (A) A left scapulo-anterior pseudoaneurysm in the left axillary area (arrow). Light microscopic images of hematoxylin and eosin–stained sections of the subclavian lymph node biopsy specimen show (B) a well-developed lymphoid follicle with eosinophilic infiltration and (C) vascular proliferation (200×).
One and half year after the diagnosis of KD, the patient presented with a month-long history of foamy urine and a weight gain of 5 kg; pitting edema of both lower extremities was also observed.
Upon admission, the patient’s blood pressure was 98/61 mmHg, white blood cell (WBC) count was 7,200/mm3 (with 33.8% eosinophils), and hemoglobin and platelet levels were 14.5 g/dL and 287,000/mm3, respectively. Serum blood urea nitrogen and creatinine levels were within the normal range, with a creatinine clearance rate of 120 mL/min/1.73 m2. Other laboratory assessments showed normal electrolyte, low serum albumin (1.3 g/dL), and high cholesterol (521 mg/dL) levels; complement proteins (C3 and C4) and immunoglobulins (Ig) A, G, and M were within normal limits. The patient’s virology profile was negative for hepatitis B and C, and her immunologic status was negative for anti-double-stranded DNA, antistreptococcal antibodies, cryoglobulins, and rheumatoid factor. Urinalysis was 4+ positive for protein, with five to six red blood cells and five to nine WBCs per high-power field. The random, spot urinary protein-to-creatinine ratio (uPCR) was 16.6 g/g, and urine protein electrophoresis showed nonspecific proteinuria.
A light microscopic examination of renal biopsy specimens showed diffuse thickening of the capillary walls, focal, mild hypercellular mesangial cells, and normal-sized glomeruli. An electron microscopic examination revealed electron-dense deposits in some subepithelial areas and diffuse foot process effacement. An immunofluorescence microscopic examination revealed granular deposits of IgM, IgG, C3, and C1q in the mesangium (Fig. 2). Overall, the MN was diagnosed as Ehrenreich and Churg Stage III. Angiotensin receptor blocker or angiotensin-converting enzyme inhibitor was considered the first-choice treatment. However, the patient’s blood pressure was too low to add antihypertensive drugs to the treatment protocol. The proteinuria slowly resolved (from 16.6 g/g/d to 2.0 g/g/d) over a 6-month period upon treatment with cyclosporine (3.5 mg/kg/d) and prednisolone (dose tapered from 0.5 mg/kg/d to 0.17 mg/kg/d within 6 months). However, 1.5 years from the onset of nephrotic syndrome, the patient’s random uPCR and serum creatinine level increased to 31.0 g/g and 2.11 mg/dL, respectively. Fractional sodium excretion was calculated to be 1.23%. Moreover, at the same time, the left subclavian mass had enlarged from an initial 5.8 cm×3.6 cm to 7.2 cm×7.2 cm, causing occlusion of the left axillary artery. Therefore, surgical resection was necessary, and pathological reports of the excised specimen were consistent with KD.
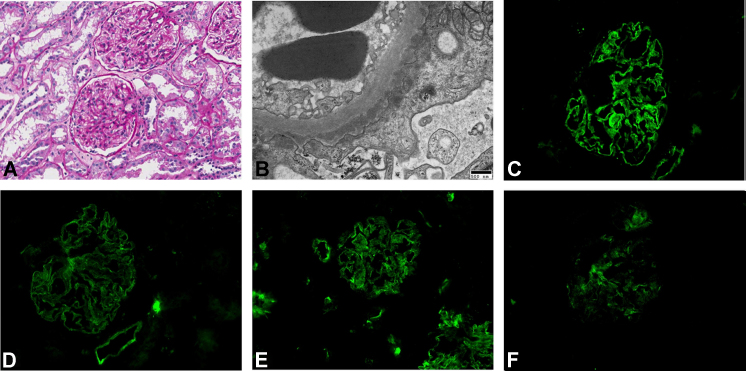
Renal biopsy specimens diagnosed with membranous nephropathy. (A) Light microscopic image of a biopsy specimen shows hypercellular mesangial cells (Hematoxylin and eosin-stained, 200×). (B) An electron microscopic image of a similar specimen reveals electron-dense deposits in some subepithelial areas and diffuse foot process effacement (20,000×). Using immunofluorescence microscopy, granular deposits of (C) IgG, 3+, (D) IgM, 2+, (E) C3, 3+, and (F) C1q, 1+ were detected in the mesangium (400×). Ig, immunoglobulin.
Fortunately, after the mass excision, the azotemia normalized from a serum creatinine level of 2.11 mg/dL to 1.11 mg/dL, and proteinuria resolved from a random uPCR of 31.0 g/g to 6.9 g/g within 1 month. After 3 months of follow up, the patient’s serum creatinine level was 0.68 mg/dL, and the random uPCR was 2.78 g/g (Fig. 3).
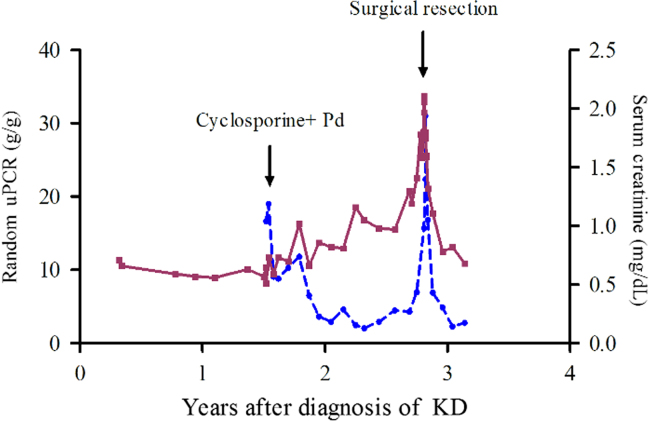
Serum creatinine and random urinary protein-to-creatinine ratio (uPCR) over time. Initial treatment with cyclosporine and prednisolone (Pd) resulted in transient proteinuria improvement. However, surgical resection of the left subclavian mass was followed by recovery of renal function. The solid line represents serum creatinine levels (mg/dL), whereas the dashed line represents the random uPCR (g/g). KD, Kimura disease.
Discussion
KD often follows a benign, chronic course but does not generally resolve spontaneously and shows frequent relapses. Although most patients do not have systemic symptoms, 12–16% of these patients have nephropathy that often presents as nephrotic syndrome [2].
Although the pathogenesis of renal involvement and vasculitis in KD is unclear, several immunopathogenetic features have been suggested. Katagiri et al [4] showed that elevated tumor necrosis factor-α, interleukin (IL)-4, IL-5, and IL-13 are found in the peripheral blood cells of KD patients, as well as in the lesion tissue. This observation supports the fact that T-helper 2 (Th2) cytokines play a part in the development of KD [5]. In addition, Lu et al [6] reported a case of KD secondary to chronic renal graft failure. Because chronic graft rejection is considered to be induced by Th2 cell-produced ILs, Th2 cytokines and overgrowth of the CD4+ Th2 may play an important role in KD.
Many treatment methods were proposed for KD, including corticosteroid, cyclosporine, surgical excision, and radiotherapy (RT). Corticosteroids were reported to be effective in controlling KD. This implies that T-cell- mediated pathogenesis maybe involved in KD since corticosteroids are known to regulate T-cell proliferation and lymphokine production. However, recurrence upon termination of steroid therapy was reported in 15–40% of the patients. Recent reports suggest that immunosuppressants such as cyclosporine and vincristine reduce recurrence rate [7], [8]. Katagiri et al [4] reported on the effectiveness of cyclosporine (4 mg/kg/d for 3 months followed by 3.5 mg/kg/d for 6 months) in treating KD. A previous study showed that RT is more effective than local excision and steroid therapy for the treatment of KD. With the 65-month median follow-up duration, local control was achieved in nine (64.3%) of the 14 patients in the RT group and in two (22.2%) of the nine patients in the non-RT group [9].
For local mass control, surgery is often recommended as the first-choice treatment [10]. However, the role of surgical resection in controlling renal manifestations in patients with KD has not yet been clarified. This case showed that surgical resection resulted in reversal of not only azotemia but also proteinuria. A similar reversal case was reported previously with regard to Castleman’s disease. A patient with a mixed type of localized Castleman’s disease complicated with nephrotic syndrome underwent removal of two mesenteric lymphoid masses. After the surgical resection, the proteinuria gradually decreased and disappeared [11]. In patients with Castleman’s disease having renal complications, immunosuppressants such as prednisolone and azathioprine are often used [12], similar to the treatment for KD. However, surgical resection is helpful to reverse the renal function.
Both KD and Castleman’s disease cases imply that some unknown circulating mediators secreted by the primary tumor may cause renal dysfunction. It has long been postulated that some circulating humoral factors might cause proteinuria in minimal change nephrotic syndrome (MCNS) and focal segmental glomerulosclerosis (FSGS) [13]. A T-cell-derived humoral factor might induce damages to the glomerular permeability barrier in MCNS. Hemopexin is one of the proposed molecules, which can cause proteinuria and glomerular alterations characteristic of MCNS [14]. In cases with FSGS, post-transplant recurrence of proteinuria and its response to plasmapheresis suggest the role of some plasma factors. Soluble urokinase receptor is detected in the plasma of patients with recurrent FSGS, and this can induce foot process effacement, proteinuria, and FSGS-like glomerulopathy [15].
In our patient, surgical mass reduction induced reversal of not only proteinuria but also the serum creatinine level. Although the patient’s serum creatinine level gradually increased over a 2-year period, the estimated glomerular filtration rate was fully normalized after surgical mass reduction. This suggests that renal dysfunction in this patient is possibly caused by a reversible mechanism. We can postulate that unknown circulating factors secreted by the angiolymphoid hyperplastic mass persistently restrict glomerular filtration possibly without evident chronic damage. Because proteinuria slowly improved compared with the serum creatinine level, some humoral factors might have induced structural change to the glomerular permeability barrier so that it took more time to recover.
We described the first official case of secondary MN in a KD patient in Korea. Moreover, renal complications in this patient were nearly normalized after surgical mass reduction. This suggests that surgical resection could be preferentially be considered prior to medical treatment in KD patients with renal complications.
Conflict of interest
All contributing authors declare no conflict of interest.