


Introduction
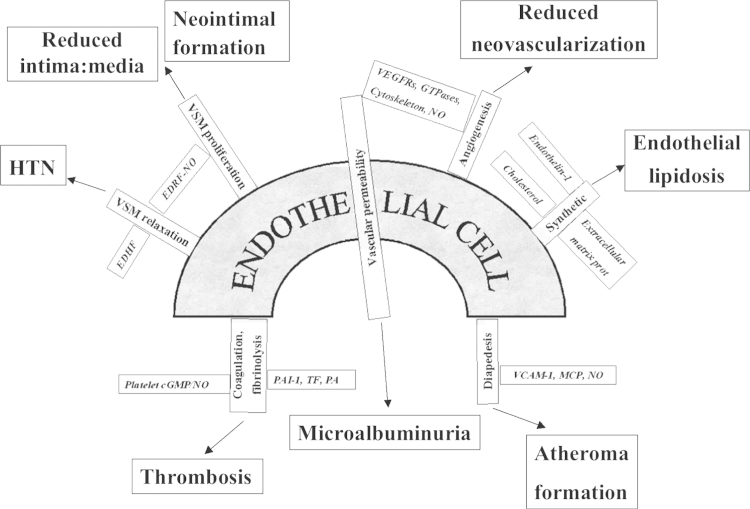
The first usage of the term ŌĆ£endothelial dysfunctionŌĆØ was related to the pulmonary circulation in bleomycin-induced pulmonary fibrosis [1]. The term received its modern meaning with the publication of a study of impaired acetylcholine-induced vasorelaxation of coronary arteries in patients with coronary atherosclerosis [2]. However, this paradoxical vasoconstrictor response to acetylcholine reflected only one of multiple functions of endothelial cells (ECs), and the term was further broadened to acknowledge the fact that in the case of EC dysfunction (ECD) abnormalities may occur in any of the functions carried out by these cells, as depicted in Fig. 1. The contribution of dysfunctional vascular endothelium to the development and progression of various diseases, including renal diseases, is difficult to overestimate [3]. However, this cell type has long been ignored by nephrologists. Only recently the attention of nephrologists has been attracted to these cells endowed with a multitude of functions, such as the regulation of hemodynamics, permeability, nutrient exchange, flux of inflammatory leukocytes, pro- and anticoagulant functions, amongst others. Therefore, dysfunction of this cell type has a myriad of consequences, as illustrated in Fig. 1.
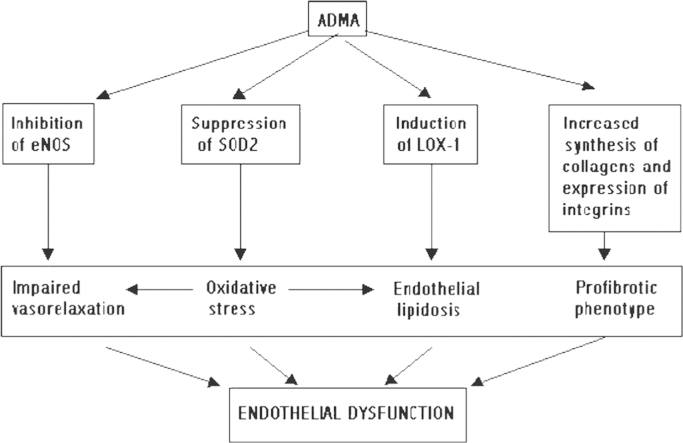
Multiple lines of evidence indicate that cardiovascular morbidity and mortality dominate the landscape of chronic kidney diseases (CKD) long before they enter the end stage. ECD is a well-known instigator of cardiovascular morbidity and it develops in CKD with a remarkable frequency. Flow-dependent relaxation of conduit vessels, a function of endothelium-derived relaxing factors, mainly nitric oxide (NO) production, is suppressed in patients and animals with CKD. Different modifications of testing endothelium-dependent vasorelaxation have become the standard for diagnosing ECD, together with other surrogate biomarkers such as markers of oxidative stress [8-iso-prostaglandin F2-alpha and oxidized low-density lipoprotein (LDL)], proinflammatory markers (high-sensitivity C-reactive protein, lipoprotein-associated phospholipase A2, soluble intercellular adhesion molecule 1, interleukin-6 von Willebrand factor), inhibitor of endothelial NO synthase (eNOS) asymmetric dimethylarginine (ADMA), circulating procoagulants, and others [4]. Diverse traditional risk factors for cardiovascular disease are joined by nontraditional risk factors, such as ADMA, advanced glycation end-products, pro-oxidants, all accumulating in CKD, conspire to induce ECD via its most reliable attribute, eNOS uncoupling, which results not only in the downregulation of NO production, but also generates oxygen free radicals. The pathogenic mechanisms of developing ECD and ensuing cardiovascular morbidity have been the focus of studies in my laboratory for the past more than two decades. Here, I would like to present a retrospective of these studies, some accomplishments in deciphering molecular mechanisms of ECD, and some inevitable shortcomings.
Screening cardiovascular-relevant genes
A cardiovascular gene screen of ECs with inhibited or uncoupled eNOS or elevated ambient concentration of homocysteine, two nontraditional mediators of ECD highly prevalent in patients with CKD, revealed that this condition is associated with the modulation of the expression of several genes, as detailed in Fig. 2, 3.
One of the pursued leads obtained from this survey was related to the upregulation of the receptor for oxidized LDL, lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) [5]. LOX-1 was induced not only in macrophages, but also in ECs cultured in the presence of NOS inhibitors. This increased LOX-1 expression was associated in ECs with cellular uptake of fluorescently labeled oxidized LDL, which was reduced two- to three-fold by coincubation with a competitive inhibitor, soluble LOX-1, and in suppression of NO production. In cultured macrophages ADMA-induced overexpression of LOX-1 was associated with accumulation of oxidized LDL and accelerated formation of ŌĆ£foamŌĆØ cells.
Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase by exposure of macro- and renal microvascular ECs to homocysteine was confirmed at the level of protein expression. Gas chromatography--mass spectrometry analyses of dysfunctional ECs revealed increased loading with cholesterol, a conditioned which we termed ŌĆ£endothelial lipidosisŌĆØ (by analogy with the hepatic lipidosis) [6]. Coapplication of a statin resulted in the normalization of cellular cholesterol and improved NO-mediated vasorelaxation.
Homocysteine-induced upregulation and redistribution of a gap-junctional protein connexin-43 (Cx-43) did not show any increase in cell-to-cell gap junctional communication. Confocal microscopy of stressed ECs expressing chimeric Cx-43 green fluorescent protein and prelabeled with mitochondrial or endoplasmic reticulum fluorescent markers revealed a profound redistribution of Cx-43 from the plasma membrane to the mitochondria. In view of the fact that Cx-43 is the main endothelial-smooth muscle cells gap junctional protein for delivery of endothelial hyperpolarizing factor, we tested nonŌĆōNO-mediated vasorelaxation in microdissected renal interlobar arteries and demonstrated the perturbed transmission of endothelium-derived hyperpolarizing factor to vascular smooth muscle cells [7].
One of the reasons the search for the mechanisms of ECD is important lies in the finding that the inhibition of NOS renders ECs to undergo endothelial-to-mesenchymal transition (Endo-MT) [8]. This in turn leads to microvascular rarefaction, elongation of distances for oxygen diffusion to tissues, and underlies the development of tissue hypoxia. These observations and conclusions were furthered by studies from Zeisberg et al [9] who examined this process in different models of CKD, and Li et al [10] who documented it in experimental type 1 diabetes. Increased synthesis of collagen XVIII [8,11] and accumulation of its antiangiogenic fragment, endostatin, were documented in vivo and in vitro after inhibition of eNOS. Endostatin treatment of ECs in culture was found to suppress the expression of endothelial markers and enhance expression of mesenchymal markers. In vivo studies confirmed actuation of Endo-MT [8], as a step toward microvascular rarefaction, tissue hypoxia, and eventual fibrosis.
All these pathways could potentially accelerate atherogenic remodeling of the vascular wall, thus contributing to the elevated cardiovascular morbidity of patients with CKD, as detailed in earlier publications [11,12]. Other findings still remain unexplored.
Proteomic abnormalities of dysfunctional ECs
ECD is associated with profound metabolic abnormalities; these not only can initiate ECD, but also tend to aggravate preexisting ECD. Addabbo et al [13] used two-dimensional electrophoresis, in-gel digestion, and mass-spectrometry analysis to screen renal microvascular isolates obtained from mice with inhibited eNOS. Data revealed 13 nonredundant differentially expressed proteins with a high level of confidence. Five of those are specific for mitochondria, and two downregulated proteins are known components of the Krebs cycle: aconitase-2 and enoyl-coA-hydratase-1 [13]. This deficiency of key enzymes of tricarboxylic acid cycle is associated with reduced mitochondrial mass, mitochondrial oxidative stress, and switch to the normoxic glycolysis (Warburg type of metabolic hypoxia seen in chronic uncoupling of eNOS) to support energy metabolism. In collaborative studies with Stoessel et al [14] we demonstrated that in this model of ECD induced by l-NG-monomethylarginine, acetate salt (l-NMMA), renal tissue hypoxia is undetectable further indicating towards normoxic glycolysis. We also observed accumulation of lactate, which confirms the fact that glycolysis is enhanced in ECD. Moreover, by supplying cultured cells with the metabolic intermediate downstream of the deficient aconitase-2-╬▒-ketoglutarate (which enters the Krebs cycle bypassing the enzymatic bottleneck) it becomes possible to restore energy metabolism and prevent cell death or premature senescence. These findings raise the question whether it could be possible to restore EC metabolism in ECD by supplementing animals with l-glutamine (GLN).
Metabolic abnormalities in ECD
Addabbo et al [15] addressed this question in the follow-up metabolomic studies of isolated renal microvasculature and plasma of mice chronically receiving an eNOS uncoupler with and without GLN supplementation. Analysis of the metabolome of renal microvasculature detected 1,400 aligned features by untargeted molecular feature extraction; 37 differential metabolites in l-NMMA+GLN versus l-NMMA and control versus l-NMMA were shared. l-NMMA-induced renal microvascular metabolite changes that were offset by GLN supplementation include myoinositol, GPC, betaine, and taurine. The latter was the only osmolyte upregulated in l-NMMA-treated mice (in contrast with the downregulation of GPC, betaine, and myoinisitol) and compensated with GLN treatment. GABA and alanine, potential inhibitors of taurine transport, were also downregulated in l-NMMA treated mice.
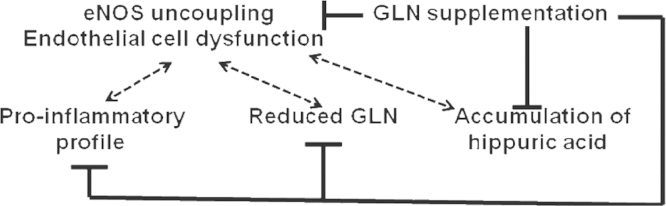
Treatment with GLN supplementation results in improved vasculopathy (as judged by restored endothelium-dependent vasorelaxation) and decreased proteinuria [15], as summarized in Fig. 4. In addition, metabolomic studies conducted using liquid chromatography--mass spectrometry analyses disclose multiple metabolite abnormalities developing in ECD and restored by GLN supplementation. Among those are lysophospholipids, hippuric acid (all elevated), and GLN/glutamate itself (reduced levels), which become normalized after GLN supplementation [15]. Hence, metabolic abnormalities affect EC functions and correcting these abnormalities leads to the amelioration of ECD and vasculopathy, both contributing to progression of CKD.
Presently, the mechanism(s) of GLN action are left for speculation. The previously described contribution of GLN to glutathione synthesis and its maintenance in reduced form by providing a source of reducing equivalents, like nicotinamide adenine dinucleotide phosphate (NADPH), is highly relevant to the present dataset. A similar effect is observed in erythrocytes, where GLN plays a role of antioxidant and preserves NADPH level required for glutathione recycling. Consistent with our previous demonstration of enhanced oxidative stress in the l-NMMA model of ECD [14], this antioxidant defense by GLN supplementation can explain some of the observed benefits of such a therapy: prevention of eNOS uncoupling and generation of peroxynitrite. An additional potential mechanism of beneficial vascular effect of GLN supplementation can be the consequence of restored Krebs cycling of intermediates with the activation of malate shuttle and enhanced production of NADPH, a cofactor of eNOS. Furthermore, the ability of GLN to be converted to l-citrulline, with the latter converted to l-arginine, may underlie the observed restoration of endothelium-dependent vasorelaxation. Such a necessity may present itself in situations accompanied by uncoupling of eNOS, as it occurs in cultured porcine aortic ECs where l-NMMA stimulated l-arginine efflux via a y+ transporter, potentially depleting its intracellular pool and further exacerbating eNOS uncoupling [15].
Another totally unexpected finding of this metabolomic analysis is related to the elevated levels of hippurate in l-NMMA-treated mice with ECD, but without uremia. Hippuric acid is a well-known ŌĆ£uremic toxinŌĆØ. In patients with CKD, the level of hippuric acid is increased from 2.2╬╝M in controls to 160╬╝M in far-advanced disease. Hippurate inhibits glucose utilization in striated muscles, thus contributing to muscle weakness, and has been suggested to accelerate the progression of CKDs. To examine the potential effects of hippuric acid on vasomotion, we employed thoracic aorta rings to study its vasoconstrictor effects and its influences on vasorelaxation. Addition of hippuric acid at concentrations of 100┬Ā┬Ąm to 1mM to the incubation bath did not produce vasoconstriction. In contrast, hippuric acid significantly and dose-dependently attenuated acetylcholine-induced relaxation of aortic rings. These findings establish an unknown action of hippuric acid, a putative ŌĆ£uremic toxinŌĆØ, to alter vasorelaxation. These observations provide a potential link between the accumulation of hippuric acid, even in preuremic animals, and the early development of vasculopathy [15].
As the renal excretion of hippurate represents the major pathway for its elimination, the finding of its elevation in our nonuremic mice displaying only mild renal impairment was rather unexpected. These findings may argue in favor of the increased metabolic rate of hippurate synthesis during syndromes associated with ECD. The above lines of investigation would require further studies.
Premature EC senescence in CKD
The role of telomere attrition and cell senescence in aging and diseased kidney has been pioneered in a series of studies performed by Melk and Halloran [16]. Studies in my laboratory [17ŌĆō20] revealed that stress-induced premature senescence (SIPS) of ECs occurs even in the presence of relatively unaffected telomeres. Diverse stress signals, such as pro-oxidants, ADMA, nonenzymatically glycation-modified proteins induce cell cycle arrest, SIPS, and eventual apoptosis in low-passage cultured cells and in young mice. Notably, SIPS can be reversed after the withdrawal of the offending stressor; however, if the stressor persists, SIPS becomes irreversible, ECs may undergo apoptosis, eventually culminating in microvascular rarefaction.
Senescent ECs not only disrupt the function of the endothelial lining of the vessels, but also affect the neighboring cells by their secretome, collectively designated as senescence-associated secretory products, which contain transforming growth factor-alpha (TGF-╬▒) galectin-3, insulin-like growth factor protein-3 (IGFBP-3),IGFBP-4 and IGFBP-6, macrophage inhibitory cytokine-1 [21]. Dysfunctional senescent ECs release collagen XVIII also, which is cleaved to yield its C-terminal antiangiogenic fragment, endostatin [8]. High-resolution mass spectrometry analysis of the secretome of endothelial progenitor cells disclosed 133 proteins, some known as membrane-bound, others known as secreted [22]. Specifically, soluble forms of vascular endothelial growth factor receptors, adhesion molecules, semaphorin 3F and TGF-╬▓, CD109, members of roundabout (robo) family, as well as endothelial markers were detected. Mass spectrometry screen of the secretome of colony-forming units, precursors of mature ECs, identified 272 nonredundant proteins of which 124 were also found in cultured endothelial progenitor cells [23]. Secretory products included matrix metallopeptidase-9, interleukin-8, migration inhibitory factor, various cathepsins and protease inhibitors, S100 proteins A11, A8 and A4, plasminogen activator inhibitor-2 and apolipoprotein E, as well as a proangiogenic and prosurvival factor, thymidine phosporylase. These investigations explain the growing interest in ŌĆ£cell-free therapyŌĆØ; several ongoing clinical trials, conducted mostly in patients with cardiovascular disease, are underway. Along the same lines of inquiry, Matsumoto et al in our laboratory used the cell-free extract of endothelial progenitor cells to treat activated fibroblasts (myofibroblasts) and discovered that this cell-free therapy prevented TGF╬▓1-induced fibroblast activation. In vivo injection of this cell-free extract subcapsularly in fibrotic kidneys prevented and reversed development of fibrosis in the mouse models of unilateral ureteral obstruction and chronic phase of folic acid-associated nephropathy (unpublished data).
The role of microRNAs in promoting organ fibrosis (fibromiRs) is rapidly expanding [24]. A list of fibromiRs includes miR-15 family, miR-21, miR-34a, miR-192, miR-199b, miR-208; whereas other miRs exhibit antifibrotic signaling, such as miR-1, miR-24, miR-29b, miR-101, miR-200b, among others.
Regeneration of microvasculature is further impaired by the production of antiangiogenic substances, like endostatin, and developing incompetence of endothelial progenitor cells. In addition, obliteration of microvasculature occurs via the process of Endo-MT, as discussed below.
Screening patients with CKD for subclinical signs of ECD
Various direct and indirect tests are used clinically to diagnose ECD. I have previously summarized some of those by reviewing individual diagnostic modalities [4]. Obviously, early preclinical diagnosis of ECD, before cardiovascular complications become overt, is the main goal of such a screening. Diverse surrogate markers of ECD have been established: levels of ADMA, homocysteine, markers of oxidative stress and inflammation, endothelial microparticles, among others, all have place in diagnosis. Functional testing of endothelial function can provide a direct unambiguous quantitative measure of the severity of ECD. Functional tests include measurement of flow-mediated vasodilation, cutaneous laser-Doppler flowmetry and imaging, measurement of aortic stiffness and pulse wave propagation, carotid intima-media thickness, and other techniques. We used laser-Doppler flowmetry and imaging to interrogate cutaneous microvasculature (which incidentally has some commonalities with renal microvasculature) in its adaptation to a short period of occlusion and thermal stimulation [25,26]. These studies demonstrated that abnormal reactive hyperemic responses to tourniquet occlusion or to thermal stimulation, so typical of patients with end-stage kidney disease with the concomitant cardiac disease or diabetes mellitus, are also observed in approximately one-half of these patients with no clinical history of cardiovascular disease or diabetes mellitus. The follow-up study compared these parameters with the risk assessment using Framingham and Cardiorisk scores. It showed that abnormal parameters of laser-Doppler flowmetry testing were associated with increased cardiovascular mortality, but Framingham and Cardiorisk scores, as well as levels of homocysteine or C-reactive protein had lesser predictive power. Collectively, these studies [25,26] re-emphasized the importance of ECD in the development of cardiovascular morbidity and mortality, and the necessity to test patients with CKD for signs of silent, preclinical ECD. Such testing is an important complement to Framingham and Cardiorisk scoring of cardiovascular risk.
Endo-MT and the role of TGF-╬▓ and endostatin signaling
Endo-MT is a physiological developmental stage occurring during embryonic formation of heart valves and septa. In adulthood, Endo-MT is a sizable contributor to vascular drop-out and development of tubulointerstitial fibrosis, as has been described in three different models of renal diseaseŌĆöunilateral ureteral obstruction, streptozotocin-induced diabetic nephropathy, and a mouse model of AlportŌĆÖs syndrome [9,10]. From 30% to 50% of interstitial fibroblasts were found to originate from the endothelium. TGF-╬▓ is a major mediator of Endo-MT, and BMP-7 is a factor counteracting Endo-MT as demonstrated using the EC fate-tracing technique. Actions of TGF-╬▓ are mediated via activin receptor-like kinases 1 and 5 (Alk1 and Alk5). Activation of Alk1 selectively expressed on ECs results in cell migration, proliferation, and angiogenesis, whereas stimulation of Alk5 induces, via Smads 2/3 phosphorylation, the transcription of SM22╬▒, fibronectin, and plasminogen activator inhibitor-1 which mediate differentiation along the smooth muscle/mesenchymal phenotype and leads to formation of myofibroblasts. The balance between activation of these two Alk pathways is regulated by endoglin, another specific endothelial TGF coreceptor. Notably, prolonged activation with TGF-╬▓ results in the escape of Alk1 signaling and predominant signaling via Alk 5, thus promoting Endo-MT [27].
Gene microarray analysis of cultured ECs treated with an inhibitor of NOS revealed upregulation of collagen XVIII and its antiangiogenic fragment endostatin, a finding confirmed in vivo in mice chronically treated with NOS inhibitor [8]. Enhanced generation of endostatin in these animals leads to the development of Endo-MT and eventual rarefaction of renal microvasculature, thus further compounding vascular and parenchymal pathology. As mentioned above, ECs exposed to diverse stressors respond with lysosomal dysfunction, leakage of cathepsins, and degradation of SIRT1. In turn, SIRT1 depletion leads to the downregulation of MMP-14 and the accumulation of ECM.
Therapeutic options
Some well-established therapeutic interventions are summarized below [28].
Inhibition of angiotensin-2 action
This is the mainstay therapy directed to slow down the progression of CKD. It uses ACE inhibitors and angiotensin receptor blockers, which exert their action through inhibition of NADPH oxidase, preservation of eNOS function, and bradykinin level, thus improving ECD.
3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors
These are important contributors to the treatment of ECD. Statins elicit their effect on ECs by reducing oxidative stress and improving eNOS function, both occurring independent of their lipid-lowering effects.
Peroxisome proliferator-activated receptor alpha agonists
Fibrates, improve EC function by reducing oxidative stress and nuclear factor kappa B activation.
Antioxidants
Tempol and ebselen, protect endothelium by preventing excessive accumulation of reactive oxygen species thus reducing eNOS uncoupling and restoring endothelial functions.
Several rational experimental strategies for vascular protection have recently emerged. These are briefly noted below.
An activator of the antioxidant Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway
This is a synthetic triterpenoid bardoxolone methyl, has been advocated as a potential therapeutic agent restoring ECD [29] and improving renal function in CKD patients with type 2 diabetes [30]. The recent ŌĆ£BEACONŌĆØ clinical trial of Bardoxolone methyl, an Nrf2 activator, however, cast some doubt in its efficacy at least in patients with type 2 diabetes and stage 4 CKD [31].
An endogenous antioxidant, lipoic acid
This may play a role in vascular and renal protection against elevated levels of angiotensin II-induced injury, as has been demonstrated in transgenic rats harboring human renin and angiotensinogen genes [32]. However, in a prospective, placebo-controlled, double-blind clinical trial of ╬▒-lipoic acid in combination with tocopherols did not influence biomarkers of inflammation and oxidative stress or the erythropoietic response in hemodialysis patients [33].
Mammalian target of rapamycin inhibitor
Rapamycin and a variety of recently synthesized more potent inhibitors, may improve ECD by preventing (SIPS) of ECs.
Activators of sirtuins
Resveratrol and newly developed analogs, may have a place among therapeutic approaches to prevent EC senescence and restore metabolic abnormalities.
GLN supplementation
This remains at the experimental stage [15], but pilot clinical trials are being initiated.
Suppression of the JNK pathway
This can be achieved through activation of adenosine monophosphate kinase by chronic treatment with 5-aminoimidazole-4-carboxamide 1-╬▓-d-ribofuranoside, Acadesine, N1-(╬▓-d-ribofuranosyl)-5-aminoimidazole-4-carboxamide or metformin [34]. Their effects are mediated via activation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha and improved mitochondrial biogenesis and cytoprotection, thus leading to attenuation of oxidative stress-induced endothelial injury.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









