


Introduction
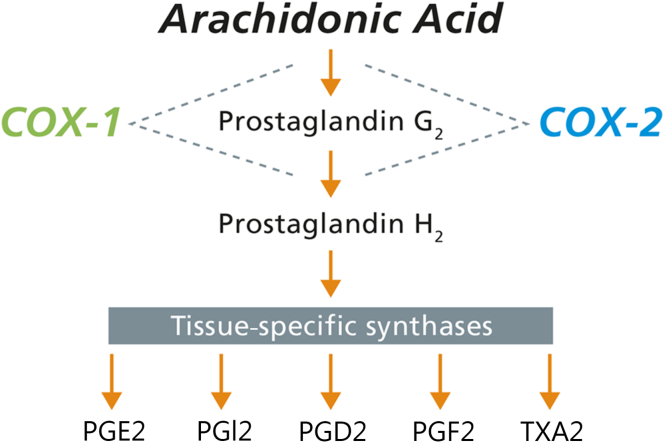
Prostaglandins (PGs) are important lipid mediators of numerous physiological and pathophysiological processes in the kidney. During normal physiological conditions, PGs play an essential role in the regulation of renal hemodynamic, renin release, as well as water and salt balance [1], [2], [3]. Likewise, PG synthesis can be stimulated in response to distinct pathophysiological situations, including inflammation, pain, and cancer [4], [5]. PGs are derived from enzymatic metabolism of free arachidonic acid to PGG2 and subsequently to PGH2 followed by the production of the bioactive PGs—PGE2, PGI2, PGD2, PGF2α—and thromboxane A2 by tissue-specific synthases (Fig. 1) [6], [7]. Cyclooxygenase (COX) is the rate-limiting enzyme that is responsible for the first 2 steps in the synthesis of PGs. The COX enzyme exists in 2 isoforms, COX-1 and COX-2. These 2 isoforms have a comparable molecular mass of 71 and 73 kDa, respectively, but their expression and distribution within the renal tissue are different. COX-1 is constitutively expressed at high levels maintaining normal housekeeping functions. COX-2 is expressed at low levels in the kidney under normal physiological conditions and is highly induced in response to inflammation and renal injury [1], [6]. In this review, we will highlight mainly the role of COX-2 and PGE2 in several physiological and pathophysiological processes in the kidney.
Physiologic conditions
COX-1 and COX-2 in the kidney
The differential expression and distribution of the 2 COX isoforms implicate them as being involved in the regulation of various physiological functions within the kidney (Fig. 2 ). COX-1 is the dominant isoform to be expressed in glomerular mesangial cells, arteriolar endothelial cells, as well as in cortical and medullary collecting ducts in the kidney of bovine, rabbit, guinea pig, rat, and mouse [8], [9]. In the human kidney, COX-1 has been identified in the collecting duct cells, interstitial cells, and vasa recta [10], [11]. In contrast, basal COX-2 expression is less intense and displays some interspecies variation in localization. COX-2 distribution has been localized in the macula densa of the cortical thick ascending limb of Henle and interstitial cells in rodents, rabbit, and dogs [8], [12], [13]. COX-2 immunoreactivity has also been described in intercalated cells of the cortical collecting ducts in mouse kidney sections [14]. In humans, COX-2 is associated with parts of the renal vasculature, loop of Henle, and podocytes [10], [11]. Furthermore, COX-2 has been detected in macula densa in humans >60 years of age [15]. In general, regarding COX-2 distribution, it is important to be aware that the previously reported expression of COX-2 has only been reported during normal physiological conditions. In response to inflammatory states, however, COX-2 may be expressed in many more cells and different cell types within the kidney [16].
COX-deficient mouse models have provided important information regarding the role of COX-2. Additional investigation into the physiological and pathophysiological effects of these 2 isoforms has also been acquired from COX-deficient mice. Importantly, mice disrupted for COX-1 appear to be generally healthy, and there are no obvious renal defects [17]. In contrast, mice with gene disruption of COX-2 have severe nephropathy, and the kidneys appear pale and smaller than those of the wild-type littermates. In the earlier states, the kidneys show small immature glomeruli in the subcapsular region with enlarged glomeruli outside of this hypoplastic area. With age, the renal pathology develops into more severe states and results in end-stage renal disease [18], [19], [20]. In addition, gender differences in renal phenotype have been reported in COX-2–disrupted mice with a male propensity for kidney injury, increased baseline water turnover, and hypertension [21], [22]. Taken together, it appears that the COX-2 isoform plays a more dominant role in the kidney diseases compared with COX-1.
PGE2 in the kidney
Although PG production occurs in all tissues, the kidney is a rich source, particularly with PGE2 being the major renal PG metabolite. PGE2 can be generated by all renal cells which are characterized by the presence of PGE2 synthases—the enzyme responsible for the production of PGE2. At present, 3 distinct types of PGE2 synthases have been identified in the kidney, including microsomal PGE synthase 1 (mPGES-1), mPGES-2, and cytosolic PGES (cPGES). The expression of mPGES-1 is inducible in response to physiological or pathophysiological stimuli and is the most abundant renal PGES form that has been considered to be functionally coupled to both COX-1 and COX-2 activity to increase the production of PGE2 [23]. In contrast, mPGES-2 and cPGES expression is constitutive rather than regulatory, which suggests that these PGESs may be housekeeping genes involved in both early and late phases of PGE2 production [23], [24]. Furthermore, mice deficient in mPGES-1, but not in mPGES-2 or cPGES, suppress PGE2 synthesis, indicating that mPGES-1 may represent an important renal pathway responsible for PGE2 production [25], [26], [27]. In addition, studies have demonstrated reduced inflammatory response in mPGES-1–deficient mice, providing evidence that mPGES-1 plays a pivotal role in the production of PGE2 linked to inflammation [28], [29].
PGE2 plays an important role in modulating the effect of vasopressin on the osmotic water reabsorption in the renal collecting duct cells where it attenuates antidiuretic action. This effect has been attributed to both inhibition of cAMP synthesis and elevation of cytosolic calcium (Ca2+) in rabbit cortical collecting ducts [30], [31] and rat terminal inner medullary collecting ducts causing decreased trafficking of aquaporin 2 (AQP2) to the apical plasma membrane [32], [33]. In addition, the signaling pathways underlying the diuretic effect of PGE2 include the cAMP and Ca2+-independent activation of the Rho-kinase and formation of F-actin [34].
The involvement of PGE2 in the regulation of body water metabolism was also demonstrated by the association between the urinary PGE2 excretion and urine concentration [35], [36]. In nephrogenic diabetes insipidus (NDI), patients have severe polyuria and urinary concentrating impairment in the presence of normal or high plasma levels of arginine vasopressin (AVP), and urinary excretion of PGE2 is also increased [35], [36]. In an animal study of lithium-induced NDI, the 24-hour urinary excretion of PGE2 was also significantly increased [37]. These observations have led to the use of COX inhibitors to concentrate urine and decrease urine volume in NDI patients [38], [39], [40]. In contrast, urinary PGE2 excretion is decreased in patients with central diabetes insipidus and can be increased by AVP treatment in parallel with increased urinary concentrating ability [36]. Similarly, PGE2 concentration in kidney tissue slices from congenitally AVP-deficient Brattleboro rats is low and can be increased in response to V2 receptor agonist 1-desamino-8-d-arginine vasopressin (dDAVP) treatment [41].
The biological actions of PGE2 are exerted by the activation of 4 G-protein–coupled prostanoid receptors, designated EP1, EP2, EP3, and EP4 [42]. Through these receptors, PGE2 plays a variety of renal physiological roles, including tubular water and sodium transport, glomerular filtration, as well as vascular resistance [43], [44]. EP1 stimulates intracellular Ca2+ and contributes to the natriuretic and diuretic effects of PGE2 [45], [46] as well as plays an important role in regulation of the blood pressure [47]. The EP2 and EP4 receptors activate adenylate cyclase and increase cAMP production, whereas the EP3 receptor inhibits cAMP signaling [42]. EP2 and EP4 can increase water reabsorption via different intracellular signaling pathways [48], whereas EP3 decreases water permeability leading to increased water excretion [46]. PGE2 or the EP1/3 agonist, sulprostone, decreases vasopressin-induced osmotic water permeability and AQP2 membrane trafficking without affecting cAMP levels or AQP2 phosphorylation at Ser256 (protein kinase A phosphorylation consensus site) [41], [49]. In a recent review, Olesen and Fenton [50] provided a detailed information about the collecting duct water permeability, highlighting the action of PGE2 and EP receptors. Although PGE2 attenuates the AVP-mediated urine concentration, a number of unanswered questions still remain regarding the role of PGE2 in body water homeostasis.
Pathophysiological conditions
COX-2 and PGE2 has been demonstrated to play key roles in the pathophysiology of a variety of renal disorders (Table 1). This section focuses on 2 major renal diseases—namely obstructive nephropathy and acute kidney injury (AKI).
COX-2 and PGE2 in obstructive nephropathy
Obstructive nephropathy is one of the most common kidney diseases, which may result in serious consequences for kidney function. Obstructive nephropathy is characterized by the impairment of most renal function. Initially, renal blood flow and glomerular filtration rate (GFR) are reduced, and in parallel, most tubular functions become impaired leading to severe reductions in the ability to handle sodium and water along the nephron. The molecular explanation for this is severe downregulation of almost all sodium transporters, cotransporters, and AQP water channels [51]. The consequences are the development of a severe urinary concentrating defect. In the more chronic phase of obstruction, renal tissue is characterized by progressive inflammation and development of tubulointerstitial fibrosis. There are numerous causes, including kidney stones, cancer of the prostate, and congenital malformations, contributing to the obstructive nephropathy [52].
The PG system is activated in response to obstructive nephropathy, and several studies have demonstrated that ureteral obstruction is associated with a marked induction of COX-2 and increased PGs and thromboxane synthesis [12], [53], [54], [55], [56], [57], [58], [59], [60], [61]. Pharmacological intervention for the treatment of obstructive nephropathy has been focused on different approaches, including the hemodynamic dysregulation, altered water and salt handling, and the development of inflammation and tubulointerstitial fibrosis. The role of PGs and thromboxane in the dysregulation of hemodynamic parameters is complex as these lipid mediators are a mixture of both vasoconstrictors and vasodilators. Specific blocking of the vasoconstrictor thromboxane further attenuates the reduced GFR in response to obstructive nephropathy, but GFR is unchanged when PG production is nonselectively inhibited by the COX inhibitor indomethacin [62].
Several studies have investigated the role of PGs on altered water and salt handling in obstructive nephropathy. It has been described that pharmacological inhibition of COX-2 prevents increased PGE2 production as well as the downregulation of renal water channels and sodium transporters in response to acute bilateral ureteral obstruction [12], [56]. In addition, we have demonstrated that disruption of COX-2 prevents downregulation of the expression of the water channel protein AQP2 and AQP3 in the cortex of the mice subjected to bilateral ureteral obstruction [54]. In the postobstructive kidney, COX-2 inhibition attenuates the downregulation of AQP2 and reduces polyuria acutely after the release of obstruction [55], [60]. Thus, the data indicate that the increased COX-2–mediated PGE2 synthesis plays an important role for the dysregulation of renal AQPs and sodium transporters, which contributes to the impaired urinary concentrating capacity in response to obstructive nephropathy.
The PG system is similarly an important pharmacological target for the prevention of renal inflammation and fibrosis. Studies have shown that selective COX-2 inhibition ameliorates the progression of renal parenchymal damage and interstitial fibrosis caused by unilateral ureteral obstruction (UUO) [63], [64]. In addition, we have inhibited COX-2 using a chitosan nanoparticle system delivering anti–COX-2 small interfering RNA (siRNA) that targets COX-2 in only macrophages. These particles have been administrated by intraperitoneal injection, and the nature of the chitosan nanoparticles and the macrophage-rich environment in the peritoneum provide avid uptake into macrophages, and these macrophages are then recruited to the obstructed kidney. The results demonstrated that siRNA knockdown of COX-2 prevents or minimizes renal damage, inflammation, and apoptosis in a UUO model, placing RNA interference (RNAi) as a new potential therapeutic in COX-2 inhibition [58]. Likewise, it has recently been observed that antioxidants can inhibit the induction of COX-2 in renal medullary interstitial cells in response to UUO, suggesting that reactive oxygen species/oxidative stress might play a role in the regulation of the COX-2–mediated progression of renal damage and inflammation [57].
Finally, the PGE2/EP4 receptors have been revealed as potential pharmacological targets in obstructive nephropathy. EP4 receptor can affect injurious responses, and Nakagawa et al [65] have demonstrated that EP4 can also limit the progression of tubulointerstitial fibrosis by suppressing the inflammatory response in response to UUO.
COX-2 and PGE2 response in AKI
AKI represents an acute reduction in renal function and a change in the structure that can lead to increased morbidity and mortality in critically ill patients [66]. Previous studies demonstrated that experimental AKI induced by complete obstruction of the renal arteries followed by reperfusion in rats revealed structural alterations in renal tubule epithelia in association with impaired urinary concentrating ability and sodium excretion [67], [68]. The impairment of urinary concentration was associated with significantly decreased expression of AQPs in the kidney collecting duct as well as in the proximal tubule [67]. Such an experimental model with complete renal artery occlusion followed by reperfusion only partly mimics AKI in human patients. More than 70% of AKI in humans is of circulatory nature and results from hypoxic injury to the kidney, including hypovolemia or hemorrhagic shock [69].
COX inhibitors, including both nonsteroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors, have been associated with an increased risk of AKI [70]. AKI could be complicated in the setting of NSAID therapy, particularly in the clinical situations of decreased effective circulating volume, including chronic kidney disease, congestive heart failure, and liver cirrhosis. Under these circumstances, a fall in effective circulating volume leads to activation of the renin–angiotensin and adrenergic nervous system, which results in renal vasoconstriction and impaired renal function. This will stimulate endogenous renal synthesis of vasodilatory PGs to antagonize the vasoconstriction and normalize renal perfusion and GFR. Therefore, an inhibition of these PG-dependent counterregulatory mechanisms will disrupt the balance between vasoconstriction and vasodilation and potentially give rise to AKI [71], [72], [73].
The application of NSAIDs or COX-2 inhibitors in patients with high-risk factors of AKI might be dependent on dose and duration of treatment, and drugs should be used with caution to reduce AKI occurrence. Huerta et al [70] demonstrated an increased risk of AKI with long-term therapy and high dose of NSAIDs compared with low-medium dose. Furthermore, they did not find a greater risk at the beginning of NSAID treatment. Therefore, is short-term and low-dose application of COX inhibitors associated with increased AKI incidence? In some forms of AKI, COX inhibitors have revealed beneficial roles in animal models. Studies have demonstrated that indomethacin, which is one of the most widely used NSAIDs in clinics, attenuates severe dysfunction as well as renal inflammation and fibrosis after ischemia/reperfusion (I/R)–induced AKI in rodents [74], [75], [76], [77], [78]. Furthermore, it has recently been shown that the protective effects of indomethacin are highly dose dependent in mice subjected to I/R injury [74]. Likewise, studies have reported that specific COX-2 blockade can minimize the progression of renal damage and oxidative stress in cisplatin- and I/R-induced AKI [77], [79]. In addition, recent studies have shown that COX-2 inhibition can prevent oxidative injury in response to renal I/R in a dose-dependent manner [80], [81]. In contrast, other studies have revealed that suppression of COX-2 by pharmacological inhibitors or COX-2 knockout mice increases renal dysfunction and injury after renal I/R [82], [83]. Ranganathan et al [84] also demonstrated that COX-2–mediated PGE2 production exacerbates I/R-induced injury via the EP4 receptor. The inconsistency between the various studies may be due to different experimental strategies such as different animal species and I/R models with different duration of ischemia or reperfusion time as well as selection of different COX-2 inhibitors. The results of COX-2 disruption could be different according to the different types of AKI, possibly due to the diversity of the pathogenic mechanisms. Taken together, the effects of NSAIDs or selective COX-2 inhibitors may be dependent on the various pathophysiological mechanisms and insults related to kidney disorders.
Conclusion
This review has provided evidence that the COX-2–derived PGs are implicated in a variety of physiological and pathophysiological processes in the kidney. During normal physiological conditions, PGs play important roles in the regulation of renal hemodynamic and homeostasis of body water and sodium balance. In addition, COX-2 and PGs are involved in different pathophysiological conditions, including obstructive nephropathy and AKI. The role of COX-2 and COX-2 inhibitors in numerous pathophysiological conditions still remains controversial as detrimental versus beneficial effects of COX-2 occur and may depend on the underlying pathophysiological conditions. This reveals a counterbalanced intrarenal handling of COX-2 highlighting the importance of the enzyme in renal physiology.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









