Current concepts of the podocyte in nephrotic syndrome
Article information
Abstract
Nephrotic syndrome is a disorder of the glomerular filtration barrier, and central to the filtration mechanism of the glomerular filtration barrier is the podocyte. We are starting to better understand how this cell, with its unique architectural features, fulfils its exact filtration properties. The multiprotein complex between adjacent podocyte foot processes, the slit diaphragm, is essential to the control of the actin cytoskeleton and cell morphology. Many of the proteins within the slit diaphragm, including nephrin, podocin, transient receptor potential-6 channel, and α-actinin-4, have been identified via genetic studies of inherited nephrotic syndromes. Signaling from slit diaphragm proteins to the actin cytoskeleton is mediated via the Rho GTPases. These are thought to be involved in the control of podocyte motility, which has been postulated as a focus of proteinuric pathways. Nephrotic syndrome is currently treated with immunosuppressive therapy, with significant adverse effects. These therapies may work in nephrotic syndrome due to specific effects on the podocytes. This review aims to describe our current understanding of the cellular pathways and molecules within the podocyte relevant to nephrotic syndrome and its treatment. With our current knowledge of the cellular biology of the podocyte, there is much hope for targeted therapies for nephrotic syndromes.
Introduction
The nephrotic syndrome (NS) describes a triad of hypoalbuminemia, edema, and proteinuria. It is the most common glomerular disease of childhood and has an incidence of approximately 2:100,000 [1].
NS may be classified by response to steroids—steroid responsive and steroid resistant. It is possible that a different pathophysiology underlies these different subgroups; however, this does not explain those who are initially responsive but later become resistant. Minimal-change nephropathy is reported as the most common cause of NS in children (approximately 80% of cases) according to the International Study of Kidney Disease in Children (1967–1974). Most of these will be steroid responsive. Approximately 20% of NS will be clinically steroid resistant [steroid-resistant NS (SRNS)]. and about 60% of these will have focal segmental glomerulosclerosis (FSGS) on biopsy. Those with FSGS may progress to end-stage renal failure requiring renal transplant. One of the greatest challenges in managing FSGS is the recurrence risk of 30–50% after the first transplant and higher for subsequent grafts [2].
The glomerular filtration barrier (GFB) is composed of two cell types, the capillary endothelial cells and the podocytes, separated by a specialized glomerular basement membrane. In addition, mesangial cells support the structure of the glomerulus, and parietal epithelial cells line the capsule encircling the glomerular capillaries (Fig. 1). The cell–cell junction between adjacent podocyte foot processes is known as the slit diaphragm and is a highly sophisticated complex of proteins that orchestrates signaling to the interior of the cell.
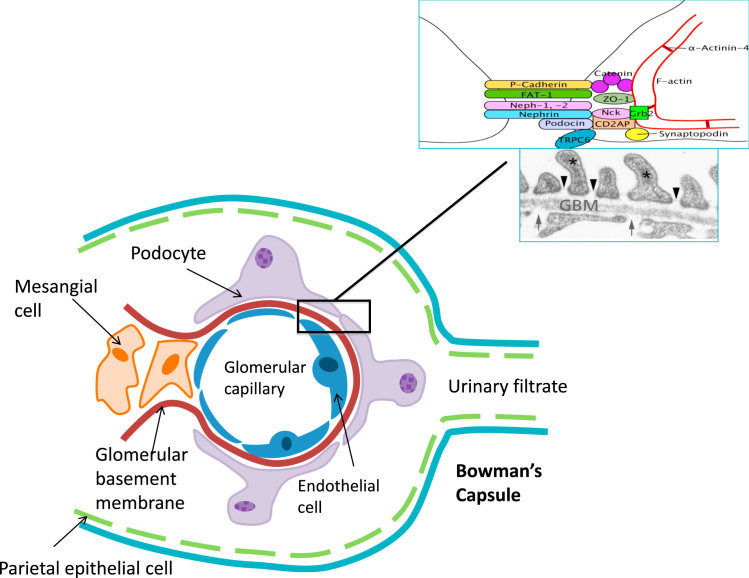
Diagram of a glomerular capillary, showing the three cell types, as well as the parietal cells of Bowman’s capsule, and their interrelationship in supporting the glomerulus and forming the filtration barrier.
Much has been discovered about the genetics of NS since the discovery of the gene encoding nephrin in 1998 [3]. Nephrin, along with other proteins in the slit diaphragm, is now known to be essential to the filtration mechanism of the GFB. Mutations in genes encoding slit diaphragm proteins have led to proteinuria and renal failure in animal models, and have also been identified in humans with NS. The discovery of these slit diaphragm proteins has led to subsequent studies on podocytes identifying both downstream pathways with effects on actin cytoskeleton remodeling within foot processes, as well as upstream factors involved in the pathogenesis of NS. In this review, we will discuss the slit diaphragm proteins, the cellular pathways in the podocyte, the control of the podocyte actin cytoskeleton, podocyte motility, and the effects of current therapies for NS on the podocyte. Finally, we will elucidate some of the current breakthroughs that are helping us to understand the nature of circulating factors in the pathogenesis of SRNS.
Podocyte physiology
The tri-layer GFB comprises of the fenestrated capillary endothelial cells, the glomerular basement membrane, and the podocyte. The podocyte has many unique properties that are key to its role in ultrafiltration.
The podocyte consists of a cell body, major processes, secondary processes, and finely interdigitating foot processes. The slit diaphragm membrane, which links the foot processes, controls the ultrafiltration of molecules by signaling to the actin cytoskeleton within the foot processes. In addition to the actin cytoskeleton within the foot processes, other important structural components of the podocyte include vimentin-rich intermediate filaments and microtubules. These components all contribute towards cell shape and rigidity. Microtubules regulate cell motility, vesicular transport, cell polarity, and organization and positioning of the membrane organelles. Crosstalk between actin and microtubules enables changes at the slit diaphragm to be transmitted to the nuclei via microtubule-associated proteins, and enables the podocyte to respond to signals from the foot processes [4].
Under the cell body and its major processes lies the subpodocyte space. This is estimated to cover 60% of the GFB, and the level of resistance within this space (which is controlled by tight exit pores) contributes significantly to the overall permeability of the GFB [5].
It is interesting that the podocyte, traditionally described as an epithelial cell, shows several features of smooth muscle cells when fully differentiated. It expresses smooth muscle cell markers such a smoothelin and calponin [6], and is thought to have a contractile function [7]. It has been speculated that, instead of it being the mesangial cell, the podocyte might have the role of supporting capillary pressures, preventing capillary expansion and maintaining ultrafiltration [6].
Regulation of the actin cytoskeleton
A universal pathological feature of NS is podocyte foot process effacement, as seen by electron microscopy. The dynamic control of the actin cytoskeleton is essential in maintaining the shape and movement of the foot processes, as well as in maintaining cell–cell contacts. More than a hundred proteins are involved in the regulation of actin, enabling processes such polymerization and depolymerization [9]. When arranged linearly as a central core of filament bundles, actin can provide significant tensile strength [8]. Actin branching in the form of filopodia or lamellipodia is likely to drive cell motility, as has been seen in the growth cones of neurons. Actin monomers can self-assemble, but this is kinetically unfavorable. The two important catalysts for rapid actin assembly are formins, which enable actin filaments to elongate at a rapid rate [10], and the actin-related protein 2/3 (Arp2/3) complex, which is activated by the Wiskott–Aldrich syndrome protein (WASP) family of proteins to form branched actin networks [9]. The Ena/VASP family of proteins have emerged as regulators of actin assembly and cell motility [11].
Rho GTPases
The small GTPases of the Rho family (RhoA, Rac1, Cdc42) are some of the most important proteins in regulating actin dynamics. For example, Rac1 can stimulate Arp2/3 and promote branched actin formation, and can also induce actin polymerization at cell–cell junctions to strengthen cell–cell contacts [12]. The Rho GTPases cycle between two distinct conformational states: they are active when bound to GTP and inactive when bound to GDP [7]. The complex interplay between these small GTPases and their tightly controlled spatial and temporal distribution play a significant role in modulating the podocyte cytoskeleton and cell–cell adhesion. RhoA has a role in the initial events of protrusion and is activated at the cell edge synchronous with edge advancement, whereas Cdc42 and Rac1 are activated just behind the cell edge 40 seconds later than RhoA, and activate pathways implicated in the reinforcement and stabilization of newly expanded protrusions [13].
When a podocyte-specific, doxycycline-inducible constitutively active form of RhoA was created in transgenic mice, it enhanced actin polymerization, decreased nephrin mRNA and protein levels, promoted podocyte apoptosis, and decreased endogenous RhoA levels [14]. Increased RhoA activity corresponded to increased levels of albuminuria and to histological changes of FSGS and extensive foot process effacement [15]. Conversely, transgenic mice with the dominant negative form displayed a loss of podocyte stress fibers, suggesting that a basal level of RhoA plays an important role in maintaining podocyte structure [14]. A low level of RhoA was associated with foot process effacement that was not evident on light microscopy, and although most mice with low levels of RhoA had low levels of albuminuria, a high level persisted in some mice [15]. It appears that low levels of RhoA may play a role in minimal-change nephropathy, but this role is not yet entirely clear.
This is just one example of how the control of the actin cytoskeleton is regulated in the podocyte, and how disruption of this regulation could lead to NS. This tight regulation involves many other proteins, many of which have been identified via studies of familial NS. Mutations in these single genes often result in SRNS and have histological changes correlating to that of FSGS. Most of these genes code for proteins within the slit diaphragm, which interact with the actin cytoskeleton by signaling through the Rho GTPases (Fig. 2).
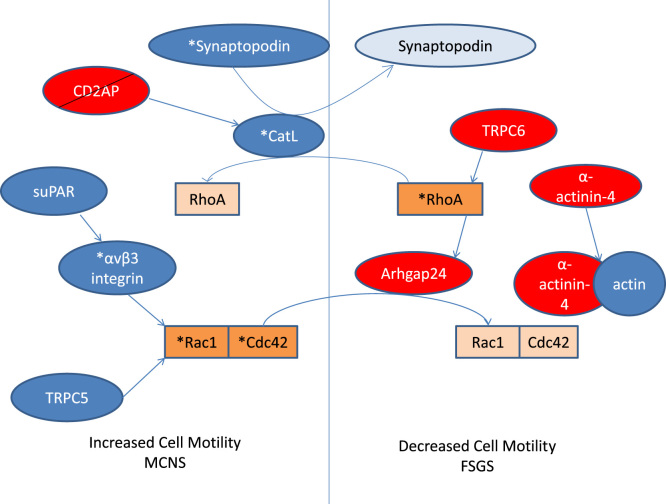
A schematic view of regulation of RhoGTPase activation, and hence control of cell motility, by slit diaphragm proteins. Asterisks represent activated forms, and lighter colored boxes inactivation or degradation. Red boxes indicate proteins associated with familial FSGS. FSGS, focal segmental glomerulosclerosis; MCNS, minimal-change nephrotic syndrome; see text for other abbreviations.
Slit diaphragm proteins
Nephrin, encoded by NPHS1, is perhaps the most important of the slit diaphragm proteins [3]. First described in 1998, mutations in this gene result in congenital NS of the Finnish type, which presents with massive proteinuria in utero and at birth. Nephrin is a transmembrane protein with eight extracellular immunoglobulin domains, a fibronectin III domain, and an intracellular domain with several tyrosine residues [16]. Phosphorylation of these tyrosine residues by Fyn allow binding of Nck adapter proteins, which interact with N-WASP or p21-activated kinases (PAKs) to regulate the actin cytoskeleton [17], [18]. PAKs, along with other molecules downstream of nephrin-like phospholipase cγ1 and phosphoinositide 3-OH kinase (PI3K), are thought to interact with Rac1 (and Cdc42 in the case of PAK) to mediate actin reorganization [19], [20]. Nephrin is also known to interact with IQ motif containing GTPase activating protein (IQGAP), an effector protein for Rac1 and Cdc42, which regulates actin via the Arp2/3 complex and formins [21].
The gene NPHS2, encoding podocin, was discovered in 2000 [22]. Mutations in NPHS2 are the most frequent cause of NS among families with congenital (39.1%) and infantile (35.3%) NS [23]. Podocin is a member of the stomatin protein family and contains a lipid recognition motif that enables it to localize to lipid rafts in the slit diaphragm. Podocin recruits and stabilizes nephrin at these lipid microdomains, and augments its activity by approximately twofold in vitro [24].
CD2-associated protein (CD2AP) is another slit diaphragm protein that has been found to be recruited by podocin to lipid rafts [25]. CD2AP-deficient mice were found to have defects in their foot processes and mesangial cell hyperplasia, and died of renal failure by 6–7 weeks of age [26]. CD2AP, like nephrin, has been shown to associate with PI3K. Both podocin and CD2AP have been shown to augment nephrin-induced activation of AKT, a downstream mediator of PI3K [27].
CD2AP is recruited by Rac1 to regulate cell–cell contacts in the podocyte [28]. Although the other Rho GTPases have not been shown to have direct interactions with CD2AP, RhoA has now been shown to be involved in a signaling pathway mediated by CD2AP [29]. An elegant study demonstrated that lack of CD2AP promoted upregulation of cytosolic cathepsin L (CatL) via translocation of dendrin to the nucleus, resulting in the proteolysis of synaptopodin, dynamin, and RhoA, which regulate the actin cytoskeleton. These cells exhibited a hypersensitivity to transforming growth factor-beta-mediated apoptosis [29].
α-Actinin-4 is an actin-bundling protein required for maintenance of the actin cytoskeleton and podocyte adhesion. Mutations in this protein have been reported in adult-onset autosomal dominant FSGS [30]. A point mutation of α-actinin-4 (K255E) associated with FSGS has been shown to disrupt actin assembly at the junctional complexes [31]. Mutations in K255E are associated with an increase in actin-binding affinity, hence enabling the formation of misfolded actin/α-actinin-4 aggregates, which diverts actin from its normal localization at stress fibers and cell–cell junctions. Of note, this mutation also confers a resistance to regulation by calcium [32]. Studies on the corresponding mouse mutant (K256E) have shown similar microaggregates, as well as increased targeting of mutant protein for degradation, impairment of the ubiquitin–proteosome system, increased endoplasmic reticulum stress, and exacerbation of apoptosis [33].
Transient receptor potential-6 channel (TRPC6) is a Ca2+-permeable nonselective cation channel at the slit diaphragm that interacts with nephrin and podocin. Mutations in TRPC6 result in late-onset autosomal dominant FSGS, which, interestingly, mostly results in a pathological increase in calcium influx [34]. Evidence exists that TRPC-mediated calcium influx increases RhoA activity and inhibits podocyte migration. The authors of this paper hypothesize that the excessive calcium influx associated with TRPC6 mutations may render the podocyte too “stiff” to respond to cues in the environment, resulting in stress fiber and cytoskeleton disorganization and cell death [35]. It has been suggested that TRPC6 may allow the slit diaphragm to detect membrane stretch and respond to this by remodeling the actin cytoskeleton to a contractile state [36].
Microtubules
As mentioned above, microtubules are another important structural component of the podocyte. Members of the formin family of proteins not only control actin dynamics, but are also involved in the remodeling of microtubules [37]. Hence formins have the unique ability of being able to directly regulate and coordinate both the actin and microtubule cytoskeletons. Mutations in inverted formin-2, a member of the formin family, have been identified as a major cause of autosomal-dominant FSGS, accounting for 17% of cases in one study [38]. Six out of seven mutations found in this study were localized to a subdomain thought to bind to IQGAP; hence the authors suggest that a disturbance in this interaction might result in disruption in the cytoskeleton [38]. Microtubules play a significant role in cell motility, due to their mechanical properties, their ability to transport proteins along their tracks, and their interaction with actin and with other proteins at the cell edge [39].
Podocyte motility
As discussed above, both microtubules and actin appear to have significant roles in coordinating not only cell shape, but also cell motility. There is increasing evidence that the regulation of cell motility may also be of significance in the prevention of proteinuria, although the degree of motility that is considered normal or pathological has yet to be defined. Using multiphoton imaging, movement was seen in a few cells in an experimental model of FSGS in rats, suggesting that motility might be present in vivo [40].
Unsurprisingly, cell motility also appears to be coordinated by the Rho GTPases. Arhgap24, which has been found to be mutated in an autosomal dominant form of FSGS, converts Rac1 and Cdc42 to an inactive state and reduces cell spreading and motility [41]. Another study showed that the urokinase receptor was required to activate αvβ3 integrin, which promotes cell motility and activation of Rac1 and Cdc42 [42]. Blockade of αvβ3 integrin reduces podocyte motility in vitro and causes proteinuria in mice [42]. The increased activity of CatL, which was upregulated in CD2AP-deficient podocytes [29], results in a hypermotile podocyte phenotype and is associated with increased proteinuria [43]. Another regulator of RhoA is synaptopodin—an actin-associated protein that binds to α-actinin-4 and CD2AP—which inhibits the degradation of RhoA and promotes cell motility [44]. TRPC6 and TRPC5 act antagonistically to mediate cell motility, the former reducing cell migration via RhoA activation, and the latter promoting cell migration via Rac1 activation [35]. It appears that there is perhaps an optimal level of RhoA that maintains an optimal degree of motility to prevent proteinuria, as the lack of RhoA results in proteinuria via a hypermotile phenotype [14], [15], [29], [43], and the excessive activation of RhoA results in a decreased migratory phenotype that may also result in proteinuria [14], [15], [35].
Effects of current therapies on the podocyte
Current therapies for NS include glucocorticoids and calcineurin inhibitors, which are thought to act via an immunosuppressive mechanism. There is now increasing evidence that these drugs actually target the podocyte, and that their efficacy in NS might in fact not only be mediated by their immune-modulating mechanisms.
Glucocorticoids have been shown to stabilize actin structures, increase RhoA activity, and enhance recovery of podocytes from puromycin aminonucleoside nephrosis in a murine model of NS [44]. Dexamethasone in human and murine podocytes augmented podocyte survival via manipulation of cell cycle regulators, for example by downregulating p53 and p21, and upregulating bcl-xL [45], [46]. Another suggested mechanism of action for dexamethasone has been in upregulation and restoration of defective nephrin transport [46], [47]. Above, we discussed the phosphorylation of tyrosine residues on the intracellular domain of nephrin as part of its signaling mechanism. Nephrin phosphorylation is decreased in puromycin aminonucleoside nephrosis and in minimal-change disease, and dexamethasone has been shown to upregulate this phosphorylation mechanism in vitro [48].
The calcineurin inhibitor ciclosporin A is another immunosuppressive agent used in NS. Its action in a model of NS has now been shown to be independent of its effect on the immune system [49]. Ciclosporin A prevents the dephosphorylation of synaptopodin, hence preventing the degradation of synaptopodin by CatL and stabilizing the actin cytoskeleton [49]. It has also been suggested that calcium signaling might mediate this pathway [50].
Rituximab is a monoclonal antibody against CD20, which is expressed on a subset of B lymphocytes. Clinically, it has been shown to be effective in reducing relapses in difficult-to-treat steroid-sensitive NS when compared with tacrolimus [51], and has been shown to reduce post-transplant proteinuria and improve glomerular filtration rate in those with recurrent FSGS undergoing kidney transplants [52]. Rituximab was designed to be highly specific for CD20, but it appears that it might have off-target effects on the podocyte. One study has suggested that this is mediated by sphingomyelin phosphodiesterase acid-like 3b (SMPDL-3b) protein, which stabilizes the actin cytoskeleton. This study showed that rituximab provided protection from SMPDL-3b downregulation in cultured podocytes, and decreased podocyte SMPDL-3b expression in post-transplant FSGS biopsies [52].
Several new therapies have shown potential as antiproteinuric drugs and have been recently discussed in detail by Mathieson [53]. One particularly interesting pathway is that of sialylation, which confers charge on certain molecules in the podocyte, such as podocalyxin. An accidental observation in mice with a homozygous mutation for uridine diphospho-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase showed reduced sialylation of podocalyxin, segmental splitting of the glomerular basement membrane, and effacement of the podocyte foot processes. The administration of N-acetylmannosamine rescued these mice with increased sialylation [54]. In another study, angiopoietin-like-4 (Angptl4), which was found to be upregulated in human and experimental NS, was deliberately overproduced in rat podocytes. This resulted in high levels of proteinuria and foot process effacement, whereas upregulation of circulating Angptl4 did not show such effects. It was noticed that the Angptl4 in NS was hyposialylated and that when N-acetylmannosamine was introduced orally, albuminuria was significantly reduced by 40% [55]. This effect is very promising and might direct the way for clinical trials.
Circulating factors and its effects on the podocyte
In SRNS, that is, FSGS, there is overwhelming evidence that a plasma circulating factor might be key to the disease process. This ranges from evidence of recurrence of proteinuria within hours of a new graft being transplanted in patients with SRNS [56], to landmark experiments on glomerular swelling after exposure to disease plasma [57].
Post-transplantation recurrence is both a clue to the pathogenesis and a problematic and distinct clinical feature. The idiopathic form of the disease reoccurs in 30–50% of cases in a newly transplanted kidney (sometimes within minutes or hours)—an event not convincingly seen to occur in the genetic forms of the disease to date. The disease can be attenuated by intensive plasma exchange in those patients. This strongly implies that circulating plasma contains elements that directly target the podocyte and disrupt cytoskeletal dynamics, and it can be reasonably implied that proteins revealed to be responsible for genetic FSGS play a key part in the unique response of podocytes to human plasma in disease.
Recently, using the burgeoning knowledge of podocyte biology detailed above and the sophisticated in vitro and in vivo tools now available [58], [59], [60], researchers have begun to approach this disease from a new perspective. Wei et al. discovered that levels of the circulating urokinase receptor suPAR are elevated in recurrent FSGS, and cause podocyte foot processes via β3 integrin in a mouse model of proteinuria [61].
We have utilized the resource of conditionally immortalized human podocytes [58] and discarded disease plasma from these patients to study altered slit diaphragm and cytoskeletal signaling that may be specific to this disease. We demonstrated a consistent response in which disease plasma relocalized the Slit Diaphragm proteins nephrin, podocin, and CD2AP to the cytoplasm, and caused enhanced cellular calcium influx [62]. Strikingly, this response was entirely dependent on the presence of nephrin, thus conferring a podocyte cell specificity on this aberrant response, and perhaps going some way to explaining why it appears that only the podocyte is affected by abnormalities in systemically circulating plasma. We went on to show that hemopexin, a circulating plasma protease previously identified as a putative “circulating factor” in NS, causes dramatic reorganization of actin in podocytes, which again is entirely nephrin dependent [63].
The question that follows these observations relates to what the downstream effects are on the cytoskeleton in response to plasma. We have recently identified phosphorylation of specific actin-regulating proteins in response to FSGS relapse plasma, compared with remission plasma, and that FSGS relapse plasma causes enhanced podocyte motility compared with relapse or control plasma (unpublished data). These disease-specific insights will help build information on how the podocyte cytoskeleton is altered in disease, as well as pointing towards specific receptors on the cell surface that may be responsible for initiating the cascade of events.
A phenomenal amount has been discovered about the molecules and pathways in the podocyte and how these are modified in both human and animal models of NS. There are now many potential targets for novel therapies to be developed for NS, which hopefully will be podocyte specific and without the adverse effects of current therapies. However, there is still much to be learnt, as it is not entirely clear how all these pathways are linked or how tightly regulated these molecules need to be in their normal nondiseased state.
Conflict of interest
No conflict of interest.