Mitochondrial dysfunction in kidney injury, inflammation, and disease: potential therapeutic approaches
Article information

Abstract
Mitochondria are energy-producing organelles that not only satisfy the high metabolic demands of the kidney but sense and respond to kidney injury-induced oxidative stress and inflammation. Kidneys are rich in mitochondria. Mitochondrial dysfunction plays a critical role in the progression of acute kidney injury and chronic kidney disease. Mitochondrial responses to specific stimuli are highly regulated and synergistically modulated by tightly interconnected processes, including mitochondrial dynamics (fission, fusion) and mitophagy. The counterbalance between these processes is essential in maintaining a healthy network of mitochondria. Recent literature suggests that alterations in mitochondrial dynamics are implicated in kidney injury and the progression of kidney diseases. A decrease in mitochondrial fusion promotes fission-induced mitochondrial fragmentation, but a reduction in mitochondrial fission produces excessive mitochondrial elongation. The removal of dysfunctional mitochondria by mitophagy is crucial for their quality control. Defective mitochondrial function disrupts cellular redox potential and can cause cell death. Mitochondrial DNA derived from damaged cells also act as damage-associated molecular patterns to recruit immune cells and the inflammatory response can further exaggerate kidney injury. This review provides a comprehensive overview of the role of mitochondrial dysfunction in acute kidney injury and chronic kidney disease. We discuss the processes that control mitochondrial stress responses to kidney injury and review recent advances in understanding the role of mitochondrial dysfunction in inflammation and tissue damage through the use of different experimental models of kidney disease. We also describe potential mitochondria-targeted therapeutic approaches.
Introduction
Renal inflammation and tissue damage during acute kidney injury (AKI) and chronic kidney disease (CKD) have been linked to mitochondrial structural and functional alterations [1,2]. Mitochondria are a highly complex interconnected network of organelles that fulfill cellular energy needs. The kidney, an organ with high energy demands, is rich in mitochondria. Mitochondrial dysfunction arising from disturbances in the regulation of the mitochondrial electron transport chain (ETC), proton gradient, and membrane potential results in reduced adenosine triphosphate (ATP) and increased production of mitochondrial-derived reactive oxygen species (mROS), which promotes kidney injury and inflammation [3,4]. Mitochondrial swelling and fragmentation and impaired mitochondrial metabolism are directly linked to the deterioration of kidney function [5]. The equilibrium between mitochondrial fusion and fission maintains healthy mitochondrial structure and function [6]. Disruption of this balance leads to mitochondrial fragmentation, loss of mitochondrial DNA (mtDNA) integrity, and cell death [3,7].
Mitochondrial fusion maintains functional mitochondria and prevents the generation of defective mtDNA [8]. Fusion is mediated through fusion proteins in the outer and inner mitochondrial membranes (OMM and IMM) called mitofusins (MFN1, MFN2) and optic atrophy 1 (OPA1), respectively [9]. MFN1 and MFN2 have distinct functions. The GTPase activity of MFN1 is higher than that of MFN2, and OPA1 fails to execute the process of mitochondrial fusion in the absence of MFN1 [9,10]. MFN2, on the other hand, regulates mitophagy [11,12]. MFN2 also facilitates tethering and interaction between mitochondria and the endoplasmic reticulum (ER) at mitochondria-associated membranes (MAMs) and maintains efficient calcium (Ca2+) uptake by mitochondria [13]. Decreased expression of mitophagy regulators [12,14] or disruption of Ca2+ signaling [15] in the kidney contributes to tubulointerstitial inflammation and kidney fibrosis.
Mitochondrial fission is controlled by dynamin-related protein 1 (DRP1), which upon recruitment to the OMM, interacts with mitochondrial fission 1 (FIS1) to facilitate mitochondrial constriction and eventual division [6-8]. During cellular injury, the activation of mitochondrial fission and concurrent arrest of mitochondrial fusion are associated with recruitment of DRP1 to the OMM and B-cell lymphoma 2 (BCL2)-associated X protein (BAX)/BCL2-antagonist/killer 1 (BAK)-mediated OMM permeabilization, which results in mitochondrial fragmentation and apoptosis [7]. Fragmented mitochondria can be removed through DRP1-regulated mitophagy to suppress cell death [16]. Mitochondrial fission thus promotes recycling of damaged mitochondria and protects healthy mitochondria from removal via mitophagy [17].
The removal of damaged mitochondria is critical to prevent renal damage caused by oxidative stress. Mitophagy, the selective autophagy of mitochondria, facilitates the recycling of damaged mitochondrial components and controls oxidative stress [18]. Mitophagy mediators include phosphatase and tensin homolog-induced kinase1 (PINK1) and Parkin (E3 ubiquitin ligase), FUN14 domain-containing protein 1, BCL2/adenovirus E1B 19kDa protein-interacting protein 3, and NIP3-like protein. The PINK1/Parkin pathway is well characterized. As a mitochondrial-targeted protein, PINK1 is imported into healthy mitochondria via translocase of the outer membrane and translocase of the inner membrane. Mitochondrial proteases then cleave the mitochondrial-targeting signal of PINK1 and translocate it back into the cytosol for degradation [18]. In damaged mitochondria with impaired membrane potential, PINK1 accumulates on the OMM and recruits cytosolic Parkin to the damaged mitochondria [19]. PINK1 can also directly phosphorylate MFN2, which further promotes the recruitment of Parkin [11,12]. Parkin ubiquitinates OMM proteins, including MFN1, MFN2, voltage-dependent anion channel, and Miro. The ubiquitinated OMM proteins, with the help of cargo receptors (p62/sequestosome 1, optineurin, nuclear domain 10 protein 52, and tax-binding protein 1) interact with microtubule-associated protein light chain 3 to promote mitophagosome formation and subsequent degradation of the cargo [19].
Thus, the balance between mitochondrial fusion/fission and mitophagy is critical for homeostasis (Fig. 1). A disruption of this balance, particularly in the kidney, which relies on normal mitochondrial function for its essential tasks, is deleterious. Emerging evidence underscores the critical role of mitochondrial dysfunction in the pathogenesis of kidney disease. This review focuses on recent advances in understanding the role of mitochondrial dysfunction in inflammation and tissue damage through various experimental models of kidney injury and on potential mitochondria-targeted therapeutic approaches to AKI and CKD.
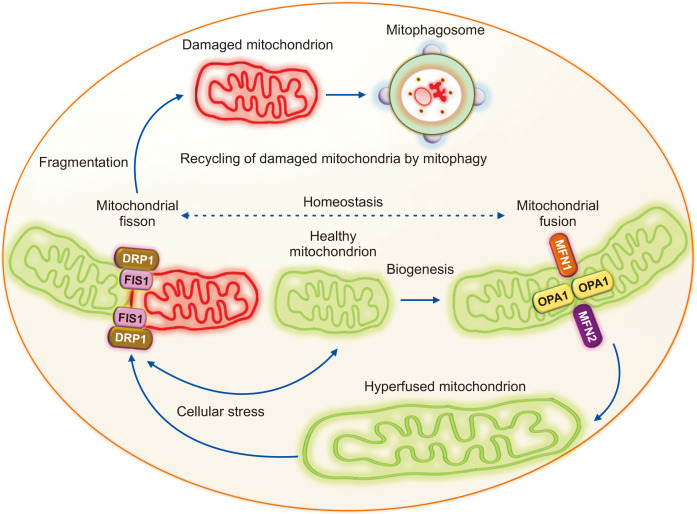
Mitochondrial fusion, fission, and mitophagy in maintaining mitochondrial homeostasis.
Balance between the opposing processes of mitochondrial fusion and fission is essential for mitochondrial homeostasis. Mitochondrial fusion is mediated by the outer mitochondrial membrane fusion proteins mitofusin 1 and 2 (MFN1 and MFN2) and the inner mitochondrial membrane fusion protein optic atrophy 1 (OPA1). The fusion of two mitochondria generates a hyperfused mitochondrion that can produce more ATP during stress. Mitochondrial fission protein dynamin-related protein 1 (DRP1) translocates to the mitochondria and binds to mitochondrial fission 1 (FIS1) to induce mitochondrial fragmentation. The damaged mitochondria with reduced membrane potential generated by mitochondrial fragmentation are then recycled via mitophagosome formation and degradation by mitophagy.
Mitochondrial dysfunction in kidney disease
The kidney has some of the highest requirements for mitochondrial content and oxygen consumption in the body, second only to the heart. The reabsorption of solutes by tubular cells is a highly ATP-demanding process, and therefore those cells have the highest mitochondrial content in the kidney [20]. Tissue damage induced by defective mitochondria during AKI is influenced by decreased ATP production, synthesis of macromolecules, an imbalanced redox state, Ca2+ homeostasis, apoptosis, necrosis, and necroptosis. Cellular fragments released by the rupture of necrotic cells or circulating mtDNA act as damage-associated molecular patterns (DAMPs), which activate innate and adaptive immune responses and the infiltration of inflammatory cells. Persistent inflammation, cell death, and defective metabolic reprogramming caused by mitochondrial dysfunction during AKI can lead to CKD (Fig. 2) and progression to end-stage renal disease (ESRD).

Role of mitochondrial structural and functional defects during acute kidney injury (AKI) and chronic kidney disease (CKD).
Mitochondrial aberrations play important roles in promoting glomerular and tubulointerstitial inflammation and fibrosis during AKI and CKD. The contributors to mitochondrial dysfunction-induced renal inflammation during AKI include: 1) activation of necroptosis via tumor necrosis factor-alpha (TNFα) and receptor-interacting protein kinase-3 (RIPK3)-mediated NADPH oxidase-4 (NOX4) recruitment to the mitochondria or through the mitochondrial permeability transition pore (MPTP), 2) mitochondrial fragmentation through increased fission and decreased fusion, and 3) damaged mitochondria-derived mitochondrial DNA (mtDNA)-mediated inflammatory response through the activation of the cyclic GMP-AMP synthase (cGAS) stimulator of interferon genes (STING) pathway. The activation of mitophagy after AKI could be a renoprotective response against mitochondrial-derived reactive oxygen species (mROS) production. Reduction in mitochondrial fatty acid oxidation (FAO), loss of cristae, and impaired mitochondrial biogenesis and decreases in mitochondrial fusion and mitophagy resulting in mitochondrial fragmentation, superoxide production, and reduced adenosine triphosphate (ATP) content all contribute to an increase in tissue damage and inflammatory and fibrotic responses in CKD.
FA, fatty acid; IFNγ, interferon gamma; IL, interleukin.
Mitochondrial dysfunction in acute kidney injury
Mitochondrial dysfunction plays a critical role in the pathogenesis of AKI and is considered an early event in various forms of AKI [21]. Here we review the evidence for the role of mitochondrial dysfunction in promoting inflammation-induced renal damage during AKI caused by sepsis, ischemia-reperfusion injury (IRI), and cisplatin-induced nephrotoxicity.
Sepsis-induced AKI
Sepsis is associated with a systemic inflammatory response to infection that can lead to multi-organ failure, including kidney failure. Patients with sepsis-associated AKI frequently have poor clinical outcomes, with high rates of morbidity and mortality [22]. Studies show that mitochondrial dysfunction contributes to the pathogenesis of sepsis-induced AKI [23]. Remarkably, evidence of mitochondrial damage in the kidney in human sepsis was reported in autopsy samples examined by electron microscopy, which found mitochondrial swelling within the tubular cells, more than four decades ago [24]. Mitochondrial structural and functional derangements in proximal tubular epithelial cells have also been reported in experimental sepsis models [5]. The inflammatory response in sepsis-induced kidney injury might be associated with the release of pro-apoptotic factors and mitochondria-derived DAMPs [25]. Moreover, the induction of apoptosis and functional alterations in podocytes and tubular cells after treatment with plasma from patients with sepsis-induced AKI suggests that circulating plasma factors augment kidney injury [26]. In the cecal ligation and puncture (CLP) model of sepsis, circulating plasma and peritoneal mtDNA and renal mROS and mitochondrial vacuolization in the proximal tubules increased after CLP [27]. The absence of toll-like receptor 9 (TLR9) reduced mtDNA-mediated kidney damage, suggesting that mtDNA induces an inflammatory response by activating TLR9. We observed increased circulating plasma and urinary mtDNA in sepsis-induced AKI [28].
Kidney biopsies from patients with sepsis displayed acute tubular necrosis, increased infiltration of immune cells, and tubular cell apoptosis [29]. Necroptosis (programmed necrosis) also exerts critical functions in the pathogenesis of sepsis-induced AKI. We reported that sepsis-induced renal tubular injury and mitochondrial dysfunction improved in the absence of the necroptosis regulator receptor-interacting protein kinase-3 (RIPK3). The mechanism through which RIPK3 induces defects in the mitochondria of renal tubular cells involves synchronized recruitment of NADPH oxidase-4 (NOX4) to the mitochondria and the suppression of mitochondrial complexes I and III [28]. NOX4 also functions as an oxygen sensor and contributes to fostering the renal inflammatory response. We observed higher urinary RIPK3 in patients with sepsis. It was further confirmed that RIPK3 inhibition reduced endotoxin-(in vivo) or lipopolysaccharide (in vitro)-induced tubular cell apoptosis [30]. The apoptotic and necroptotic pathways of cell death converge at the mitochondria. Tumor necrosis factor-alpha (TNFα), a key mediator of the sepsis-induced inflammatory response, promotes necroptosis by translocating the RIPK1/RIPK3/MLKL complex to the mitochondrial membrane [31]. However, TNF-dependent necroptosis also occurs after mitochondrial depletion through the overexpression of Parkin [32]. Therefore, the sepsis-associated renal inflammatory response and renal damage could be modulated by correcting mitochondrial structural and functional aberrations.
Ischemia-reperfusion injury
Ischemic kidney injury is a major cause of AKI. Decreased total mitochondrial content in the proximal tubular epithelial cells after IRI suggests mitochondrial damage [33]. Mitochondrial swelling, loss of cristae, and impaired IMM potential in renal tubular cells and podocytes after IRI worsen oxidative stress and renal inflammation [34]. Hypoxia-induced mitochondrial fragmentation, permeabilization of the OMM, ATP depletion, and apoptosis of proximal tubular cells after IRI have also been reported [35]. DRP1 mediates mitochondrial fission in response to ischemia/ATP depletion, resulting in renal tubular mitochondrial fragmentation, IRI-induced cell death, inflammation, and reduced kidney function [36]. The proximal tubule-specific deletion of Drp1 protects against tubular apoptosis and inflammation after IRI, suggesting that the absence of mitochondrial fission might shift the mitochondrial dynamics toward fusion for cytoprotection [36]. Under IRI or ATP depletion, DRP1 is dephosphorylated at serine (Ser)-637, which promotes DRP1 translocation to the OMM, mitochondrial fragmentation, and subsequent renal tubular cell apoptosis [37]. These results suggest that DRP1-regulated mitochondrial dynamics are an important mediator of AKI.
Mammalian STE20-like kinase 1 (MST1) is a regulator of mitochondrial fission via DRP1 phosphorylation at Ser-616 and enhancement of the F-actin assembly that induces mitochondrial injury, cytochrome c release, and mROS production, thereby promoting tubular cell apoptosis in IRI, which was attenuated by genetic deletion of Mst1 [38]. In human ischemic kidneys with IRI from cadaveric kidney transplants, the duration-dependent increase in tubular apoptosis is directly related to the release of cytochrome c from the mitochondria [39]. Further evidence suggests that mitochondria play key roles in apoptosis in IRI. BH3 interacting-domain death agonist (BID) is a proapoptotic member of the BCL2 family that targets mitochondria, and Bid knockout mice display reduced mitochondrial damage, tubular cell apoptosis, and improved renal function and survival after IRI, compared with controls [40]. BID activates BAX and BAK to regulate mitochondrial dynamics [41]. In healthy cells, BAX and BAK support MFN2, promote mitochondrial fusion, and regulate mitochondrial morphogenesis [42]. Under pathological conditions, BAX and BAK promote mitochondrial fragmentation by the sumoylation of DRP1 and its association with the OMM [43]. Therefore, BID might cause mitochondrial damage in IRI through the induction of mitochondrial fragmentation and apoptosis.
The inflammatory microenvironment in the kidney after IRI favors T-helper 1 and natural-killer T cell-derived interferon gamma (IFNγ) to promote the differentiation of monocytes into M1/proinflammatory macrophages [44]. Mitophagy exerts protective functions against ischemia-induced renal inflammation, and the absence of Pink1 and Park2 (encoding Parkin) induces ischemia-mediated mitochondrial damage, mROS, and kidney injury [14]. Furthermore, the renal cytoprotection conferred in proximal tubular cells by ischemic preconditioning occurs by clearing damaged mitochondria through the activation of PINK1-dependent mitophagy [45]. These findings suggest that the disruption of renal mitochondrial dynamics after IRI exaggerates apoptosis, oxidative stress, and inflammation to induce renal damage. Imbalance in mitochondrial fusion and fission and the suppression of mitophagy thus promote ischemia-mediated kidney injury.
Cisplatin-induced AKI
A major side effect of cisplatin chemotherapy is nephrotoxicity, which causes renal tubular cell injury with mitochondrial structural and functional impairments, particularly in the proximal tubular epithelial cells, which are rich in mitochondria. Following renal uptake, cisplatin in the cytoplasm is hydrolyzed into a positively charged electrophile that can accumulate within the negatively charged mitochondrial membrane and disrupt its membrane potential [46]. This, in turn, induces superoxide generation by negatively affecting mitochondrial metabolism, the efficiency of the ETC, and ATP production [47]. Cisplatin-induced nephrotoxicity is also associated with increased DRP1-mediated mitochondrial fragmentation, cytochrome c release, apoptosis of proximal tubular cells, and renal injury [38]. Furthermore, blockade of mitochondrial fission abrogated cisplatin-induced proximal tubular cell apoptosis and renal injury and improved kidney function, further attesting to the critical role of mitochondrial dynamics in AKI [35].
Renal inflammation and tissue damage during cisplatin-induced AKI are mediated by the innate immune response. The cyclic GMP-AMP synthase (cGAS) stimulator of interferon genes (STING) pathway induces innate immunity through the recognition of pathogen-associated molecular patterns or DAMPs, which triggers a type I IFN-mediated inflammatory response [1]. Cisplatin-induced mtDNA release into the cytosol activates the cGAS-STING pathway, with subsequent inflammation and kidney injury, and STING knockdown ameliorates the inflammatory response [1]. Evidence also reveals a direct involvement of the Fas receptor (FasR)- and TNF receptor 1 (TNFR1)-mediated apoptotic pathways in cisplatin-induced renal tubular cell death and kidney dysfunction [48]. Inhibition of death receptor-mediated pathways by genetic deletion of Tnfr1 and FasR in renal epithelial cells resulted in dramatically reduced cell death after cisplatin treatment [48]. Similar to IRI-induced AKI, cisplatin activates the pro-apoptotic protein BAX, which accumulates in the mitochondria and induces the permeabilization of the OMM, cytochrome c release, and renal tubular apoptosis [49]. Bax knockout mice had a lower number of apoptotic cells, attenuated cytochrome c release, and improved kidney function compared with controls [49]. Moreover, cisplatin induces depletion of myeloid cell leukemia-1, a pro-survival member of the BCL2 family, and the associated activation of BAX, both of which play a critical role in the release of apoptosis-inducing factor from the mitochondria [50].
An ultrastructural analysis of the kidney after cisplatin treatment revealed reduced mitochondrial mass, disrupted cristae, mitochondrial swelling in the proximal tubules, and decreased cytochrome c oxidase enzyme activity [51] and mitochondrial respiratory complexes I to IV [52], suggesting that cisplatin negatively regulates mitochondrial functions. Cisplatin-induced mitochondrial dysfunction is associated with decreased proximal tubular cell respiration and oxidative phosphorylation and induces apoptosis via protein kinase Cα and extracellular-signal-regulated kinase 1/2 [53], which cause a decline in the mitochondrial membrane potential and Ca2+ uptake and increase oxidative damage to mitochondrial lipids and proteins [54]. The administration of mitochondrial-targeted antioxidants mitigated cisplatin-induced mitochondrial structural alterations, renal inflammation, and tubular injury and apoptosis and improved renal function [55]. These findings indicate the impairment of mitochondrial bioenergetics and highlight the role of mitochondrial functional aberrations in activating the innate immune response and cisplatin-induced kidney damage.
Mitochondrial dysfunction in chronic kidney disease
Emerging evidence suggests that mitochondrial dysfunction is also a key contributor to the pathogenesis of CKD. Mitochondrial structural and functional impairments are increasingly recognized as critical mediators of many kidney diseases [12,20,56]. Here, we summarize recent advances in unraveling the functional significance of mitochondrial dysfunction in promoting inflammation and fibrotic responses in the pathogenesis of tubulointerstitial fibrosis (TIF) and various forms of CKD, including diabetic nephropathy (DN), IgA nephropathy (IgAN), membranous nephropathy (MN), and polycystic kidney disease (PKD).
Tubulointerstitial fibrosis
The development of TIF is the final histopathological consequence of most CKD. Aberrant kidney repair following AKI results in renal fibrosis and progression to CKD. Mitochondrial protective strategies have been shown to suppress the proinflammatory cytokines interleukin (IL)-18 and IL-1β and nod‐like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation to ameliorate TIF and progression to CKD following prolonged ischemia [57]. Previous studies indicate that mitochondrial dysfunction plays a role in inflammation, TIF, and CKD. Genome-wide transcriptome-based analyses revealed that human fibrotic kidneys have lower expression of various mitochondrial enzymes and regulators of fatty acid oxidation (FAO) and higher intracellular lipid deposition than controls [58]. We recently reported the protective role of mitophagy during kidney fibrosis using experimental murine models of unilateral ureteral obstruction (UUO) and an adenine diet [12]. Decreased renal expression of mitophagy regulators (PINK1, MFN2, and Parkin) in experimental and human kidney fibrosis was associated with higher circulating monocyte chemoattractant protein 1 (MCP-1), a chemokine that promotes the infiltration of monocytes, inflammation, and TIF [12]. Mitophagy impairment led to an accumulation of abnormal mitochondria, augmented macrophage-induced fibrotic response, superoxide production, and reduced ATP synthesis [12]. Compromised mitochondrial bioenergetics contribute to the development of TIF and CKD [3]. In line with our findings, others have shown that deficiency of mitophagy by Pink1 or Park2 gene deletion markedly increased mROS production and mitochondrial damage, which worsened renal fibrosis after UUO [16]. These effects were rescued by a mitochondria-targeted antioxidant. Defective mitochondrial metabolism and reduced expression of mitophagy regulators have been shown to enhance the renal inflammatory and fibrotic responses and mediate the progression of CKD.
Cigarette smoke/chronic obstructive pulmonary disease-associated CKD
Recent evidence from both human and animal studies has demonstrated a previously under-recognized association between chronic obstructive pulmonary disease (COPD) and kidney injury and fibrosis, along with implications of mitochondrial dysfunction [59]. Microalbuminuria and kidney sections showing glomerulosclerosis and TIF were observed in a higher percentage of patients with COPD compared with control subjects [60]. Using a mouse model of experimental COPD induced by chronic cigarette smoke exposure, we reported evidence of mitochondrial damage, with mitochondrial swelling and loss of cristae in renal tubular cells, increased numbers of proinflammatory and profibrotic renal macrophages, and TIF [61]. Moreover, the association of urinary mtDNA with an increased respiratory burden in patients with COPD also suggests the presence of renal mitochondrial damage [62]. The amount of cellular mtDNA correlates with the expression of mitochondrial transcription factor A (TFAM), which is an important regulator of the mitochondrial genome. The loss of TFAM in renal tubular cells has also been reported in an animal model and patients with kidney fibrosis [2].
Diabetic nephropathy
Mitochondrial dysfunction is increasingly recognized as a chief cause of the development and progression of diabetic kidney disease (DKD) [63,64]. Evidence of damaged and swollen renal mitochondria has been documented in experimental and human DN [63,65]. The progression of diabetic kidney injury is associated with decreased mitochondrial membrane potential and increased mROS in renal cells, including the proximal tubule, endothelial cells, and podocytes [63]. An optimal level of mROS generation is essential for mitochondrial biogenesis, but levels that exceed local antioxidant capacity are associated with mitochondrial dysfunction and damage to mtDNA and disrupt mitochondrial metabolism in diabetic kidneys [63]. Previous studies implied that patients with DN have impaired mitochondrial metabolism and that diabetic kidneys have global suppression of mitochondrial activity [66]. A recent study found that increased urinary cell-free mtDNA correlated with decreased intra-renal mtDNA, which was associated with severe interstitial fibrosis and a low baseline estimated glomerular filtration rate (eGFR) in patients with DN [67]. However, it is uncertain whether increased urinary mtDNA occurs due to ongoing destruction of intra-renal mitochondria and intra-renal mitochondrial depletion or to renal clearance of circulating mtDNA because blood levels of mtDNA are elevated in patients with DN [66]. Nevertheless, findings showing a correlation between urinary mtDNA and kidney pathology suggest that urinary mtDNA could serve as a marker of renal mitochondrial damage and fibrosis in DN.
Advanced glycation end products (AGEs), which are nonenzymatically modified proteins, and receptors for AGEs (RAGEs) generate ROS and are known contributors to the pathogenesis of DKD. Damaged mitochondria generate excess mitochondrial superoxide, and glycation of mitochondrial proteins also contributes to mROS generation in DKD [68]. Pharmacologic inhibition of AGE-RAGE-induced mitochondrial permeability transition abrogated mitochondrial superoxide production. Moreover, RAGE deficiency prevented diabetes-induced increases in renal mitochondrial superoxide, renal cortical apoptosis, and progression of DN in mice [68].
The suppression of mitochondrial-derived superoxide in diabetic mice protected against renal tubular injury, which is mediated by mitophagy via PINK1/Parkin, and decreased the levels of mitochondrial fission proteins (DRP1 and FIS1), resulting in reduced oxidative stress and the amelioration of mitochondrial fragmentation and apoptosis [56]. Podocyte-specific deletion of Drp1 decreased albuminuria, improved podocyte morphology and mitochondrial structural defects, and prevented mesangial matrix expansion in diabetic mice [69]. In addition, Drp1-null podocytes from diabetic mice displayed improved mitochondrial fitness, oxygen consumption rates, and ATP production [69]. Increases in the number of dysfunctional mitochondria caused by an imbalance in fusion/fission and defective mitophagy in diabetic kidneys augment oxidative stress and the progression of DKD. Thus, maintenance of mitochondrial quality control appears to be critical in protecting against DKD.
IgA nephropathy
IgAN is the most common glomerulonephritis and a leading cause of CKD that can progress to ESRD [70]. Kidney biopsy from a patient with IgAN showed mild focal segmental mesangial proliferation with dominant mesangial IgA deposition and an increased number of abnormal mitochondria in the proximal tubular cells [71]. Elevated urinary mtDNA levels were observed in patients with IgAN, and changes in urinary mtDNA were inversely correlated with eGFR [72]. Whole mitochondrial genome sequencing data from patients with IgAN who were renal transplant recipients revealed an association between five common single-nucleotide polymorphisms and ESRD, suggesting that mitochondrial defects play a potential role in the progression of CKD [73]. Interestingly, higher expression and interaction between the mitochondrial protein induced in high glucose-1 (IHG-1) and cold shock protein Y-box binding protein 1 are associated with renal inflammation, TIF, and glomerulosclerosis in IgAN [74]. These findings suggest that defects in the mitochondrial genome and functions play a critical role in worsening glomerular inflammation and disease progression.
Membranous nephropathy
MN, a leading cause of nephrotic syndrome in adults, is characterized by the deposition of immune complexes between podocytes and the glomerular basement membrane. The phospholipase A2 receptor (PLA2R) has been identified as the major autoantigen on podocytes in primary MN, and thrombospondin type I domain-containing 7A (THSD7A) is a minor antigen [75]. Autoantibodies to PLA2R and THSD7A are considered to be predominantly of the IgG4 subclass [75]. Increased glomerular mitochondrial fission proteins, DRP1, phosphorylated-DRP1 (Ser-616), and FIS1, were observed in patients with MN [76]. Furthermore, cultured podocytes exhibited mitochondrial fragmentation, loss of membrane potential, and mROS production after exposure to sera from patients with IgG4-related MN compared with controls [77]. These observations provide evidence that mitochondrial dysfunction might contribute to podocyte injury and support a role for mitochondrial-dependent autoimmunity in MN [77]. Intriguingly, a study using gene expression analyses found that loss of Drp1 was associated with alterations in the expression of a large number of genes involved in immune response, including those regulated by mitochondrial fission [78]. Those findings imply that mitochondrial dynamics might play an essential role in modulating the renal immune milieu in MN.
Polycystic kidney disease
Autosomal dominant PKD (ADPKD), which is more common than autosomal recessive PKD (ARPKD), is caused by mutations in PKD1 and PKD2, which encode polycystin 1 (PC1) and PC2, respectively [79]. The PC1-PC2 complex modulates mitochondrial Ca2+ uptake at MAMs to directly regulate oxidative phosphorylation and indirectly affect mitochondrial function by maintaining the mtDNA copy number and mitochondrial morphology [79]. Therefore, mutations in PKD1 and PKD2 lead to mitochondrial dysfunction and metabolic imbalance. Additionally, non-genetic factors have been linked with pathogenesis, including proinflammatory cytokine TNFα promoting renal cystogenesis [80], increased MCP-1 in cyst-lining cells and urinary MCP-1 excretion in human ADPKD [81], and renal profibrotic macrophages in experimental ARPKD [82], which might be associated with defects in mitophagy [12]. A recent investigation showed that loss of PC2 enhanced mitochondrial Ca2+ uptake, mitochondrial bioenergetics, and mitochondrial-ER tethering associated with increased MFN2, and knockdown of Mfn2 rescued ER-dependent mitochondrial Ca2+ signaling and reduced cyst proliferation [83]. Thus, PC2 regulates mitochondrial function through MFN2.
The mitochondria of cyst-lining cells in the kidney of a mouse model of ADPKD displayed morphological abnormalities and decreased mtDNA copy numbers and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), which is a key regulator of mitochondrial biogenesis [84]. Human ADPKD cyst-derived cells also exhibited morphological and functional abnormalities and increased mROS production [84]. Those studies provide further evidence that mitochondrial dysfunction plays a functional role in cystogenesis. Moreover, kidney cysts in mouse and human ADPKD have increased renal expression of miR-17, which represses oxidative phosphorylation and FAO by inhibiting peroxisome proliferator activated receptor alpha (PPARα), a regulator of lipid metabolism [85]. Several lines of evidence support the idea that aerobic glycolysis plays a key role in PKD, along with other metabolic dysfunctions such as an increase in the pentose phosphate pathway and fatty acid synthesis and a decrease in FAO and oxidative phosphorylation [86], suggesting that a profound metabolic reprogramming occurs in PKD. Although the precise mechanisms underlying these alterations require further investigation, they offer a great opportunity to design novel therapeutic interventions for PKD, such as mitochondria-targeting strategies.
Mitochondria-specific therapeutic approaches in kidney disease
Most mitochondrial-specific therapeutic approaches that attenuate kidney damage aim to improve mitochondrial metabolism or target mROS to relieve oxidative stress-related renal inflammation. Here we discuss several mitochondrial-targeted therapeutic approaches and their targets and effects in various experimental models of AKI and CKD (Table 1) [16,34,35,56,57,63,84,87-98].

Mitochondrial-targeted therapeutics in experimental models of AKI and CKD
Mitochondria-targeted therapeutic approaches to AKI
Several mitochondria-targeted antioxidants have been explored as potential therapies in experimental models of AKI. Mito-TEMPO is a mitochondria-targeted superoxide dismutase mimetic shown to reverse renal mitochondrial dysfunction and attenuate CLP-mediated, sepsis-induced AKI, resulting in improved renal microcirculation and GFR and increased survival [87]. Mitoquinone (MitoQ) is a mitochondrial-targeted coenzyme Q that acts as an mROS scavenger and contains a positively charged lipophilic cation to increase its mitochondrial uptake. MitoQ has been used extensively in animal models and two Phase II clinical trials in Parkinson's disease and hepatitis C and shown to be safe and well-tolerated [88]. Treatment with MitoQ or SkQR1, a plastoquinone-containing mitochondria-targeted antioxidant, in a renal IRI model helped reduce mROS production and lipid oxidation and improved renal function [88,89].
Mitochonic acid 5 (MA-5) is a synthetic indole acetic acid derivative that binds the IMM protein mitofilin and facilitates mitochondrial ATP production independently of oxidative phosphorylation and the ETC [90]. MA-5 treatment before IRI enhanced ATP production, ameliorated tubular necrosis, and improved renal function [90]. Another investigational drug that targets the IMM is Bendavia, a tetrapeptide that suppressed the mitochondrial permeability transition pore (MPTP) and attenuated oxidative stress, tubular injury, and apoptosis in experimental models of IRI [91]. Mitochondrial division inhibitor 1 (Mdivi-1), an inhibitor of DRP1-mediated mitochondrial fission, partially ameliorated ischemia-induced kidney injury [35], but it worsened renal fibrosis induced by UUO, raising concerns about its use [16]. Szeto-Schiller (SS)-31 peptide (H-D-Arg-Dmt-Lys-Phe-NH2) is another mROS scavenger that selectively targets the IMM and binds cardiolipin [34]. SS-31 ameliorates ischemia-induced mitochondrial swelling and loss of cristae, reduces mROS production and tubular and endothelial cell death [34], normalizes ATP content, and improves renal function in polymicrobial-induced sepsis [92]. Activators of PGC-1α, a key regulator of mitochondrial biogenesis, prevented the progression of IRI-induced kidney injury and improved renal function [93].
It is critical to understand the molecule(s), function(s), and pathway(s) involved in mitochondrial structural and functional impairments during AKI. A better understanding of mitochondrial dynamics and bioenergetics could help to advance our understanding of AKI pathogenesis and potential therapeutic options that target mitochondria to prevent progression to CKD.
Mitochondria-targeted therapeutic approaches to CKD
Mitochondria-targeted antioxidants have also been explored for the treatment of CKD. In a mouse model of type 1 diabetes, MitoQ treatment reduced albuminuria and attenuated both interstitial fibrosis and glomerular damage [94]. MitoQ, by partially restoring mitophagy and reducing mitochondrial fission protein (DRP1) while enhancing mitochondrial fusion protein (MFN2), reduced glomerular hypertrophy, mesangial matrix expansion, and mitochondrial and tubular damage in DKD [56]. MitoQ also inhibited cyst formation in an ADPKD mouse model [84]. MitoQ is being investigated in a Phase IV controlled, double-blind clinical trial in patients with stage 3-5 CKD (ClinicalTrials.gov Identifier:NCT02364648). Another mitochondria-targeted antioxidant, Mito-TEMPO, has been shown to rescue a UUO-induced increase in mROS, collagen deposition, and TIF in Pink1 or Park2 knockout mice [16].
SS-31 has been examined as a therapy for CKD and found to suppress transforming growth factor-beta (TGF-β), tubular apoptosis, and TIF in a UUO-induced renal fibrosis model [95]. It also attenuated interstitial inflammation and renal fibrosis 4 weeks after ischemia [34] and arrested the development of glomerulosclerosis and TIF, reduced the expression of inflammatory markers, and restored glomerular capillaries, podocyte structure, and mitochondrial integrity [57]. Furthermore, SS-31 prevented tubulointerstitial inflammation, mesangial matrix expansion, glomerulosclerosis, loss of podocytes and endothelial cells, lipotoxicity, macrophage infiltration, and renal fibrosis in a mouse model of a high-fat diet and low-dose streptozotocin [96]. Fenofibrate is a PPARα activator shown to improve mitochondrial fatty acid β-oxidation and renal function in patients with DN [97]. Activators of PGC-1α that target mitochondrial biogenesis include the AMP-activated protein kinase activator AICAR (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside), which has been shown to mitigate the accumulation of lipid droplets and mitochondrial fatty acid biosynthesis in a high-fat diet mouse model [98]. Stimulators of PGC-1α enhanced mitochondrial respiration in proximal tubular cells, increased the mtDNA content in the renal cortex [99], and prevented age-related kidney damage [100] and TIF in a UUO model [58]. Curcumin, thought to act as an antioxidant, attenuated renal inflammation and injury by negatively regulating pathways involved in DKD, such as the NLRP3 inflammasome, angiotensin-converting enzyme, and nuclear factor erythroid 2-related factor 2 [63].
Future research
Recent advances strongly suggest that mitochondrial dysfunction plays a role in the pathogenesis of kidney diseases. The molecular mechanisms of the dynamics associated with structural and functional mitochondrial defects in kidney injury require further elucidation. Future research to clarify the mechanisms of mitochondrial fusion/fission and mitochondrial quality control through mitophagy in kidney diseases is highly warranted, with the hope that preclinical studies can be translated to meet the urgent clinical need for new therapeutics with a more focused approach to rectify mitochondrial defects and mitochondrial quality control with minimal unwanted side effects.
Summary
The maintenance of healthy mitochondria is especially important in the kidney, where these organelles regulate the metabolic status of a cell and persistently sense and efficiently respond to multiple stimuli and stresses. Oxidative stress derived from damaged mitochondria has deleterious effects on tissue health and contributes to the progression of kidney disease. Mitochondrial turnover, via synthesis and the recycling of damaged mitochondria through mitophagy, is essential. Additionally, the balance between fission and fusion is essential in maintaining appropriate mitochondrial size, shape, and number. Overall, mitochondria are an attractive therapeutic target for controlling renal inflammation, cell death, tissue damage, and the progression of kidney disease.
Notes
Conflicts of interest
The spouse of Mary E. Choi is a cofounder and shareholder and serves on the Scientific Advisory Board of Proterris, Inc. The remaining authors have no conflicts of interest to declare.
Funding
Mary E. Choi is supported by National Institute of Health (NIH) grants R01 HL133801, R01 HL132198, and R01 HL055330.
Authors’ contributions
Divya Bhatia and Mary E. Choi contributed to the formation of overall concept. Divya Bhatia drafted the manuscript with the assistance of Allyson Capili. Divya Bhatia and Mary E. Choi edited and revised the manuscript. All authors read and approved the final version of the manuscript.