Paricalcitol attenuates lipopolysaccharide-induced inflammation and apoptosis in proximal tubular cells through the prostaglandin E2 receptor EP4
Article information
Abstract
Background
Vitamin D is considered to exert a protective effect on various renal diseases but its underlying molecular mechanism remains poorly understood. This study aimed to determine whether paricalcitol attenuates inflammation and apoptosis during lipopolysaccharide (LPS)-induced renal proximal tubular cell injury through the prostaglandin E2 (PGE2) receptor EP4.
Methods
Human renal tubular epithelial (HK-2) cells were pretreated with paricalcitol (2 ng/mL) for 1 hour and exposed to LPS (1 μg/mL). The effects of paricalcitol pretreatment in relation to an EP4 blockade using AH-23848 or EP4 small interfering RNA (siRNA) were investigated.
Results
The expression of cyclooxygenase-2, PGE2, and EP4 were significantly increased in LPS-exposed HK-2 cells treated with paricalcitol compared with cells exposed to LPS only. Paricalcitol prevented cell death induced by LPS exposure, and the cotreatment of AH-23848 or EP4 siRNA offset these cell-protective effects. The phosphorylation and nuclear translocation of p65 nuclear factor-kappaB (NF-κB) were decreased and the phosphorylation of Akt was increased in LPS-exposed cells with paricalcitol treatment. AH-23848 or EP4 siRNA inhibited the suppressive effects of paricalcitol on p65 NF-κB nuclear translocation and the activation of Akt. The production of proinflammatory cytokines and the number of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells were attenuated by paricalcitol in LPS exposed HK-2 cells. The cotreatment with an EP4 antagonist abolished these anti-inflammatory and antiapoptotic effects.
Conclusion
EP4 plays a pivotal role in anti-inflammatory and antiapoptotic effects through Akt and NF-κB signaling after paricalcitol pretreatment in LPS-induced renal proximal tubule cell injury.
Introduction
Sepsis is a severely deregulated systemic inflammatory response to infection and predisposes the development of multiple organ failure. Acute kidney injury (AKI) is a frequent complication of sepsis and remains a serious cause of mortality in critically ill patients [1]. Cytokine production and the infiltration of inflammatory cells into the kidney are observed in cases of septic AKI and trigger cellular dysfunction, detachment, and apoptotic cell death in proximal tubules [2,3]. In addition, tubular apoptosis around inflamed lesions increases the inflammatory reaction leading to the acceleration of tissue injury [4,5]. Therefore, the prevention of inflammation and apoptosis in proximal tubule cells is critical in ameliorating the septic AKI.
Many previous studies have shown that vitamin D protects kidneys from damage in various experimental models of renal disease [6,7]. We previously demonstrated that paricalcitol has a renoprotective effect in renal ischemia reperfusion injury via the upregulation of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2) [8]. The biological actions conducted by PGE2 includes binding to the receptor subtypes EP1 to EP4 [9], and the activation of EP4 signaling particularly controls the transcriptional activity of nuclear factor-kappaB (NF-κB) [10]. EP4 signaling also activates the phosphatidylinositol 3-kinase (PI3K) pathway leading to Akt activation [11,12]. However, it remains unclear whether the protective effects of paricalcitol on proximal tubule cells in septic AKI involve the EP4-signaling pathway.
Endotoxin, as a Gram-negative bacterial lipopolysaccharide (LPS), has been widely used in establishing sepsis models for its simplicity of use and quantification [4]. Therefore, we investigated the EP4-mediated role of paricalcitol in LPS-exposed human proximal tubular epithelial (HK-2) cells. This study aimed to determine whether paricalcitol modulates the PGE2–EP4-signaling pathway in HK-2 cells and the EP4-dependent pathway is critical for the anti-inflammatory and antiapoptotic effects of paricalcitol in HK-2 cells with LPS injury.
Methods
Human proximal tubular cell experiments
HK-2 cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin. To stimulate with LPS (Sigma-Aldrich, St. Louis, MO, USA), 80% confluent HK-2 cells were incubated with serum-free DMEM for 16 hours. HK-2 cells were pre-treated with paricalcitol (19-nor-1,25-dihydroxyvitamin D2, 2 ng/mL; Abbott Laboratories, Chicago, IL, USA) for 1 hour, and then exposed to LPS (1 μg/mL). Cells were harvested 24 hours after LPS exposure to assess the effects on the expression of the vitamin D receptor (VDR), COX-2, and PGE2 receptor subtypes. The cells were also harvested for analysis of p65 NF-κB phosphorylation and nuclear translocation and Akt phosphorylation 15 minutes after LPS exposure.
Treatment of EP4 antagonist and EP4 small-interfering RNA
The EP4 blockade was induced by AH-23848 (2 μM; Santa Cruz Biotechnology, Santa Cruz, CA, USA), an EP4-specific antagonist, or EP4 small interfering RNA (siRNA) (Bioneer Corp., Daejeon, Korea). AH-23848 (2 μM) was dissolved in NaHCO3 and diluted with distilled water. AH-23848 was simultaneously pretreated with paricalcitol for 1 hour before LPS injury. siRNAs targeted to EP4 or scrambled siRNA (Bioneer Corp.) were complexed with Lipofectamine RNAiMax (Invitrogen Biotechnology, Carlsbad, CA, USA) according to the manufacturer’s instructions. HK-2 cells were transfected with EP4 siRNAs or scrambled siRNA for 24 hours. Twenty-four hours after transfection, cells were treated with paricalcitol and exposed to LPS.
Measurement of cell viability
A commercially available 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (EZ-Cytox; Daeil Lab Service, Seoul, Korea) was used for the measurement of cell viability in HK-2 cells after 24 hours of LPS treatment according to the manufacturer’s instructions. The optical densities of the samples were quantified at 450 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
Immunoblot analyses of HK-2 cells
We conducted immunoblot analyses of HK-2 cell lysates as described previously [8]. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellu-lose membranes, and incubated with antibodies against the following proteins: VDR (Santa Cruz Biotechnology); COX-2, Akt, pS473 Akt, pp65 NF-κB and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, Beverly, MA, USA); EP4 and p65 NF-κB (Abcam, Cambridge, UK). The membranes were then incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG (Invitrogen Biotechnology) and signal densities were detected and quantified using the ChemiDoc XRS Image system (Bio-Rad Laboratories).
Fractionation of the cellular membrane and cytoplasm of HK-2 cells
Fractionation of the cellular membrane and cytoplasm of HK-2 cells was performed using a Mem-PER Plus Membrane Protein Extraction kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. HK-2 cells were washed with Cell Wash Solution, resuspended in permeabilization buffer, and incubated for 10 minutes with constant mixing at 4°C. The cytoplasmic supernatant was harvested after centrifugation at 16,000g for 15 minutes. The pellet was resuspended in solubilization buffer and the solubilized membrane fraction was harvested after centrifugation for 30 minutes at 4°C.
Enzyme immunoassay
HK-2 cells were homogenized by PRO-PREPTM Protein Extraction Solution (iNtRON Biotechnology, Seongnam, Korea) and then centrifuged for 10 minutes at 12,000g. Equal amounts of protein were analyzed by PGE2 enzyme-linked immunosorbent assay (ELISA) (Cayman Chemical, Ann Arbor, MI, USA) kit. The optical densities of the samples were quantified at 450 nm using a microplate reader (Bio-Rad Laboratories).
TUNEL assay
The number of apoptotic cells was counted in the HK-2 cells by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) using a TUNEL Apoptosis Detection kit (Intergen, Purchase, NY, USA) following the manufacturer’s protocol. The immunofluorescence images for TUNEL assay were captured by confocal microscopy (LSM5 Live Configuration Variotwo VRGB; Zeiss, Oberkochen, Germany). The number of positive cells was quantified per high power field and at least 4–6 fields were analyzed for each slide.
Immunoprecipitation for EP4
HK-2 cells were lysed with kinase buffer (KB; 50 mM Tris-HCl [pH 7.4], 25 mM NaF, 40 mM β-glycerol phosphate [pH 7.4], 120 mM NaCl, 1% NP40) and then 1 mg of lysate was immunoprecipitated using 1 μg of anti-EP4 antibody (Santa Cruz Biotechnology) and protein G Sepharose 4 Fast Flow (GE Healthcare, Uppsala, Sweden). After washing with KB without 1% NP40, immunoblotting was performed with a PGE2 antibody (Biorbyt Ltd., Cambridge, UK).
Real-time reverse transcription polymerase chain reaction of HK-2 cells
Total RNA was isolated from HK-2 cells using a NucleoSpin® RNA II kit (Macherey–Nagel, Düren, Germany). cDNA was synthesized using Reverse Transcriptase Pre-mix (Elpis Biotech, Daejeon, Korea) and amplified in a Power SYBR® Green polymerase chain reaction (PCR) Master Mix (Applied Biosystems, Carlsbad, CA, USA) with gene-specific primer pairs (Table 1). The expression levels of mRNA were normalized to the expression of GAPDH. Quantitative real-time PCR was performed on an ABI 7500 FAST instrument (Applied Biosystems).

Primer sequences for real-time PCR
Statistical analysis
Data are expressed as the mean ± standard error of mean. Multiple comparisons were performed using one-way analysis of variance and Tukey’s post hoc test. All analyses were performed using SPSS software (ver. 20.0; IBM Corp., Armonk, NY, USA). A P < 0.05 was considered statistically significant.
Results
Paricalcitol upregulates COX-2 and PGE2 expression in LPS-exposed HK-2 cells
Fig. 1 shows the effects of paricalcitol on VDR, COX-2, and PGE2 in LPS-exposed HK-2 cells. Paricalcitol pre-treatment significantly increased the expression of VDR and LPS exposure caused the suppression of VDR. Paricalcitol pretreatment largely restored the expression of VDR in LPS-exposed HK-2 cells (Fig. 1A). The expression of COX-2 was increased in paricalcitol-pretreated HK-2 cells and further enhanced in LPS-exposed cells treated with paricalcitol (Fig. 1B). HK-2 cells treated with paricalcitol produced a significantly higher level of PGE2 than control cells. LPS exposure alone increased PGE2 expression than control cells and paricalcitol further enhanced PGE2 expression in LPS-exposed cells (Fig. 1C).
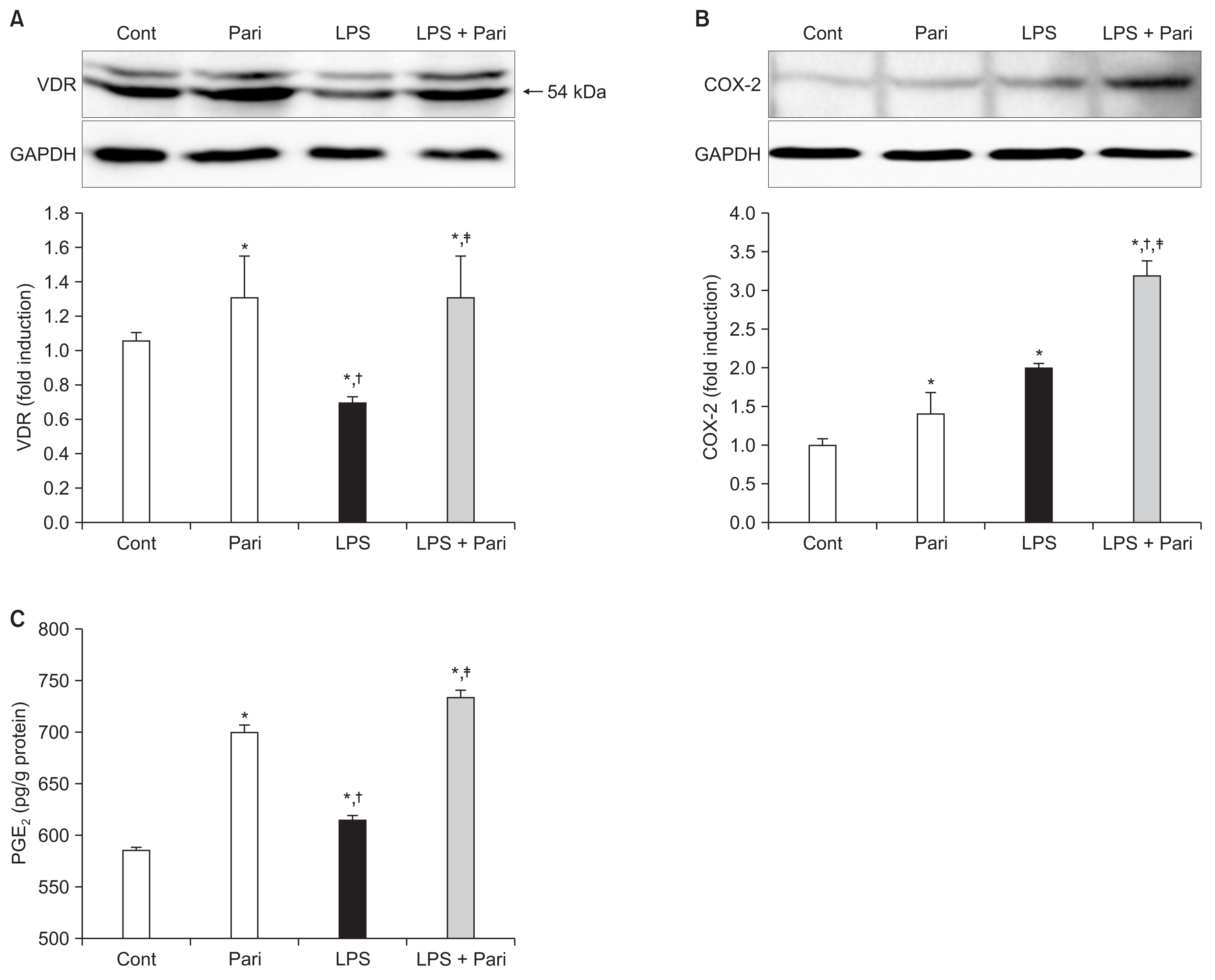
Paricalcitol upregulates the VDR, COX-2 and PGE2 expression in LPS-exposed HK-2 cells
Semiquantitative immunoblotting revealed that paricalcitol significantly increased (A) VDR and (B) COX-2 expression in LPS-exposed HK-2 cells. (C) The PGE2 level was significantly elevated in paricalcitol-treated cells compared to control cells, and further enhanced in LPS-exposed cells with paricalcitol pretreatment.
Cont, control; COX-2, cyclooxygenase-2; LPS, lipopolysaccharide; Pari, paricalcitol; PGE2, prostaglandin E2; VDR, vitamin D receptor.
Each column represents the mean ± standard error of mean of three independent experiments. *P < 0.05 vs. Cont group; †P < 0.05 vs. Pari group; ‡P < 0.05 vs. LPS group.
Paricalcitol activates the PGE2-EP4 signaling pathway in LPS-exposed HK-2 cells
We examined the induction of PGE2 receptor subtypes in paricalcitol-treated HK-2 cells. Paricalcitol significantly increased the mRNA level of EP4 in HK-2 cells with or without LPS exposure. However, paricalcitol did not affect mRNA levels of EP1–EP3 (Fig. 2A). Paricalcitol increased the expression of EP4 protein dose-dependently, and the highest increase in EP4 expression was observed at 2 ng/mL paricalcitol (Fig. 2B). The cellular membrane/ cytoplasm ratio of EP4 was significantly increased in LPS-exposed cells compared with the control group, and it was similarly increased in response to TNF-α treatment (2 ng/mL) instead of LPS (Supplementary fig. 1). Membrane/cytoplasm ratio of EP4 was further enhanced when paricalcitol-pretreated cells were exposed LPS injury (Fig. 2C). The interaction between PGE2 and EP4 was investigated by immunoprecipitation. PGE2/EP4 complex formation was increased after LPS exposure, and paricalcitol pretreatment further increased their interactions (Fig. 2D).

Paricalcitol activates PGE2-EP4 signaling pathway in LPS-exposed HK-2 cells
(A) Paricalcitol increased the mRNA level of EP4 in LPS-exposed HK-2 cells, but did not affect mRNA expression of EP1–EP3 between groups. (B) The expression of EP4 protein determined by semiquantitative immunoblot analysis was significantly increased by paricalcitol in a dose-dependent manner compared to untreated control. (C) The cellular membrane fraction of EP4 protein expression was significantly increased in LPS-exposed HK-2 cells, and was further increased by paricalcitol. (D) Immunoprecipitation confirmed that the interactions between PGE2 and EP4 was increased in LPS-exposed HK-2 cells and was further enhanced after paricalcitol pretreatment.
Cont, control; LPS, lipopolysaccharide; Pari, paricalcitol.
Each column represents the mean ± standard error of mean of three independent experiments. *P < 0.05 vs. Cont group; †P < 0.05 vs. Pari group; ‡P < 0.05 vs. LPS group.
Paricalcitol improves cell survival after LPS injury via EP4
The viability of cells treated with 0.5% NaHCO3 was similar to that of control cells without any treatment (P > 0.05). LPS treatment decreased the cell viability to around 80% of the control group, and EP4 siRNA treatment further decreased the viability of LPS-exposed cells (86.9 ± 1.7 vs. 81.7 ± 1.1; P = 0.031). Paricalcitol pretreatment significantly increased the viability of LPS-exposed cells, and this protective effect of paricalcitol was offset by AH-23848 cotreatment or an EP4 siRNA blockade (Fig. 3).
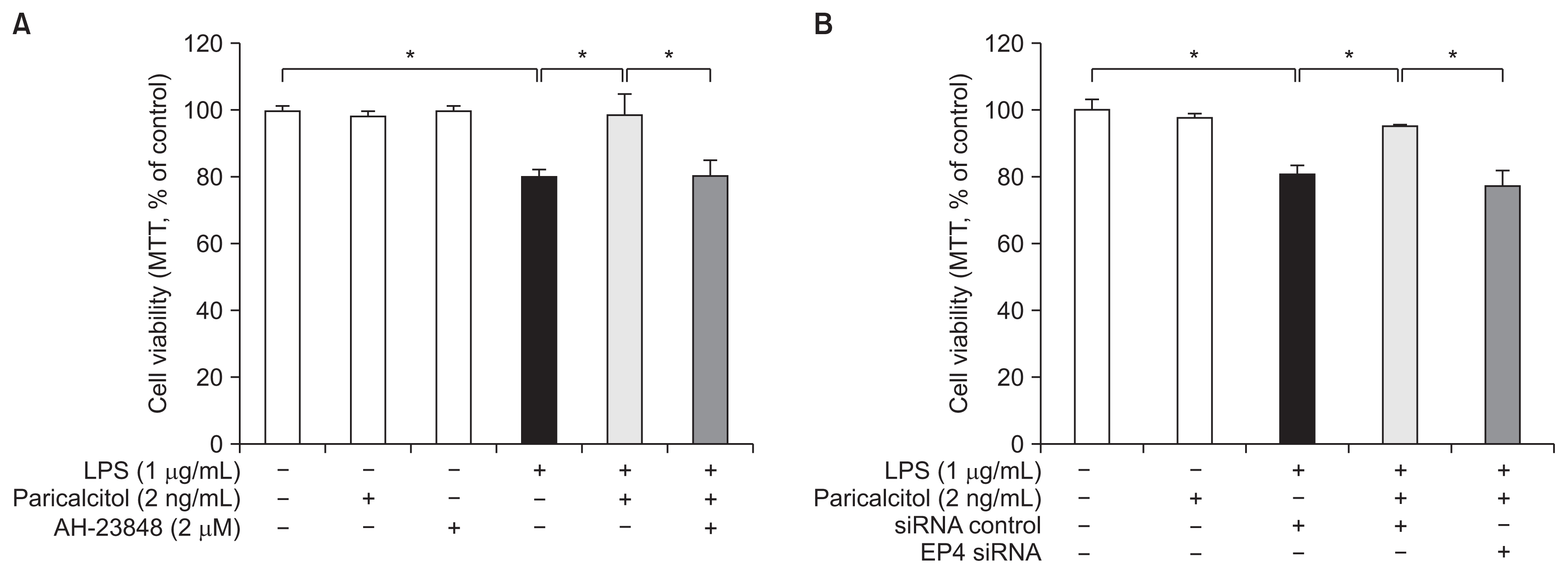
Paricalcitol protects HK-2 cells against endotoxin-induced death via an EP4-dependent pathway
Paricalcitol significantly prevented endotoxin-induced cell death. Co-treatment with (A) AH-23848, an EP4 antagonist or (B) EP4 siRNA restored the cell death to such an extent from LPS-exposed cells without paricalcitol.
LPS, lipopolysaccharide; MTT assay, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay; siRNA, small interfering RNA. Each column represents the mean ± standard error of mean of three independent experiments. *P < 0.05.
EP4 regulates the inhibitory effects of paricalcitol on p65 NF-κB activation
LPS stimulation increased the phosphorylation of p65 NF-κB (Fig. 4A). Paricalcitol suppressed phosphorylation of p65 NF-κB, and the inhibitory effect of paricalcitol was reversed by AH-23848 cotreatment in LPS-exposed HK-2 cells. We further examined the subcellular localization of p65 NF-κB (Fig. 4B). p65 NF-κB was translocated to the nuclei in response to LPS stimulation. Paricalcitol decreased p65 NF-κB translocation, and cotreatment with AH-23848 and paricalcitol promoted the nuclear translocation of p65 NF-κB in LPS-exposed cells. To evaluate whether phosphorylation of p65 NF-κB was associated with the specific activation of EP4, we performed further experiments for expression using EP4 siRNAs (Fig. 4C). As with AH-23848 treatment, EP4 siRNA enhanced the nuclear translocation of p65 NF-κB in LPS-stimulated cells treated with paricalcitol.

Paricalcitol inhibits p65 NF-κB activation after LPS treatment via an EP4-dependent pathway
p65 NF-κB phosphorylation and nuclear translocation was examined by semiquantitative immunoblot analysis. (A) Paricalcitol prevented phosphorylation of p65 NF-κB in LPS-treated cells. This inhibitory effect was reversed by AH-23848 co-treatment. *P < 0.05. (B) p65 NF-κB translocated to the nuclei in response to LPS stimulation. This translocation was suppressed by paricalcitol, whereas AH-23848 co-treatment blocked the suppressive effects of paricalcitol. *P < 0.05. (C) EP4-specific siRNA enhanced the nuclear translocation of NF-κB in LPS-exposed cells treated with paricalcitol.
Cont, control; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; Pari, paricalcitol; siCont, control siRNA; siRNA, small interfering RNA. Each column represents the mean ± standard error of mean of three independent experiments. *P < 0.05 vs. Cont group; †P < 0.05 vs. LPS + siCont group; ‡P < 0.05 vs. LPS + Pari + siCont group.
The EP4-dependent pathway contributes to the inhibitory effects of paricalcitol on inflammatory cytokine production
We investigated mRNA levels of proinflammatory cytokines, such as regulated upon activation normal T cell expressed and secreted (RANTES), interleukin-1β (IL-1β), monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α). Induction of RANTES, IL-1β, MCP-1 and TNF-α mRNA was greater in LPS-exposed HK-2 cells than controls. Paricalcitol significantly reduced the production of these proinflammatory cytokines and EP4 siRNA largely reversed this decrease (Fig. 5).
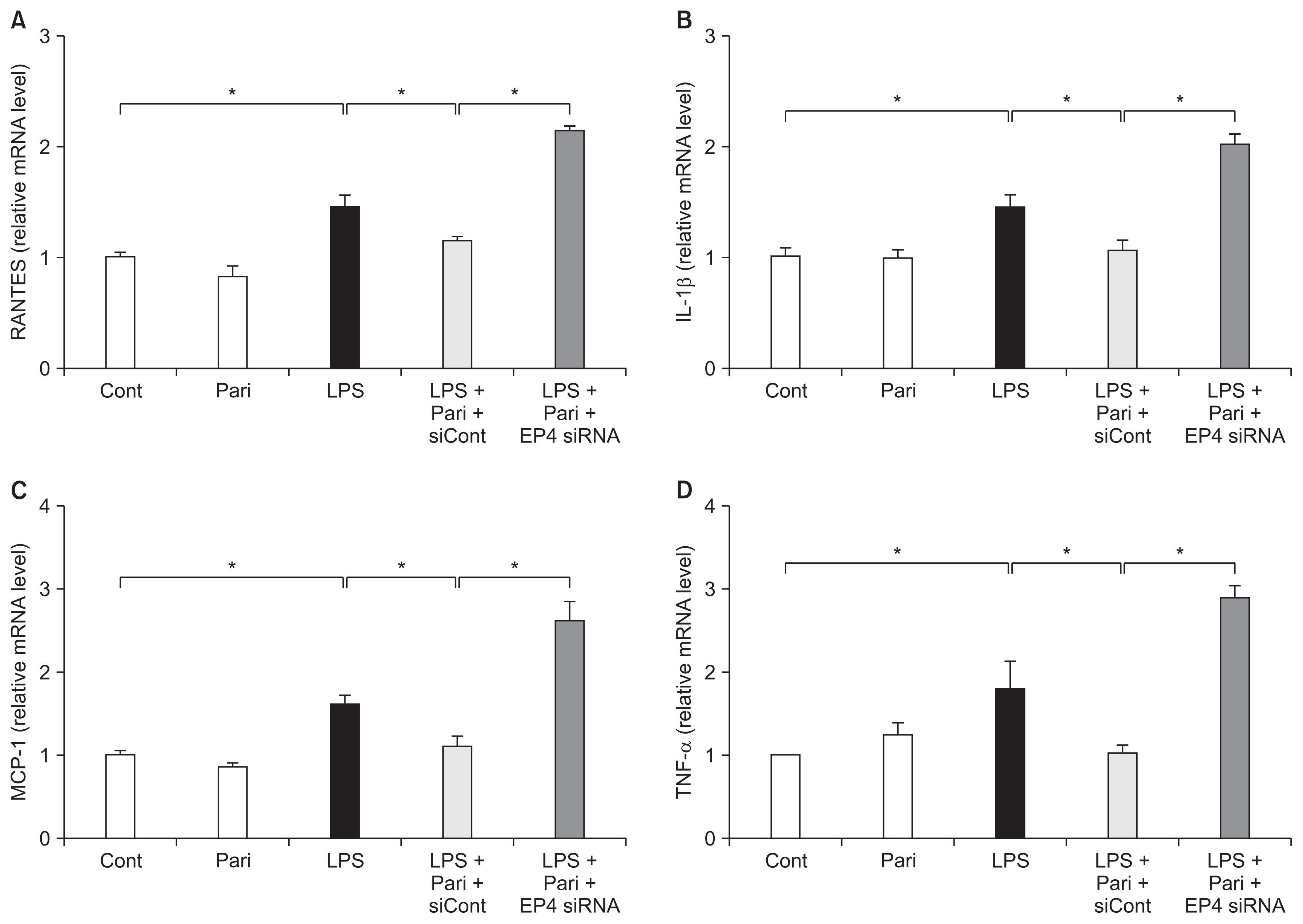
Paricalcitol suppresses the expression of inflammatory cytokines via an EP4-dependent pathway in LPS-exposed HK-2 cells
RT-PCR analysis showed that the mRNA levels of (A) RANTES, (B) IL-1β, (C) MCP-1 and (D) TNF-α were elevated in LPS-exposed HK-2 cells. Paricalcitol suppressed the LPS-induced overexpression of these inflammatory cytokines and EP4-specific siRNA ameliorated all of these suppressive effects.
Cont, control; IL-1β, interleukin-1β; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein-1; Pari, paricalcitol; RANTES, regulated upon activation normal T cell expressed and secreted; RT-PCR, real time polymerase chain reaction; siCont, control siRNA; siRNA, small interfering RNA; TNF-α, tumor necrosis factor-α. *P < 0.05.
Paricalcitol increases phosphorylation of Akt via EP4 in LPS-exposed HK-2 cells
Akt phosphorylation was significantly increased in response to endotoxin administration. Paricalcitol further enhanced the phosphorylation of Akt in LPS-exposed cells, whereas cotreatment with AH-23848 decreased phosphorylation of Akt (Fig. 6A). Similarly, EP4 siRNA significantly inhibited the phosphorylation of Akt in LPS-exposed cells treated with paricalcitol (Fig. 6B).
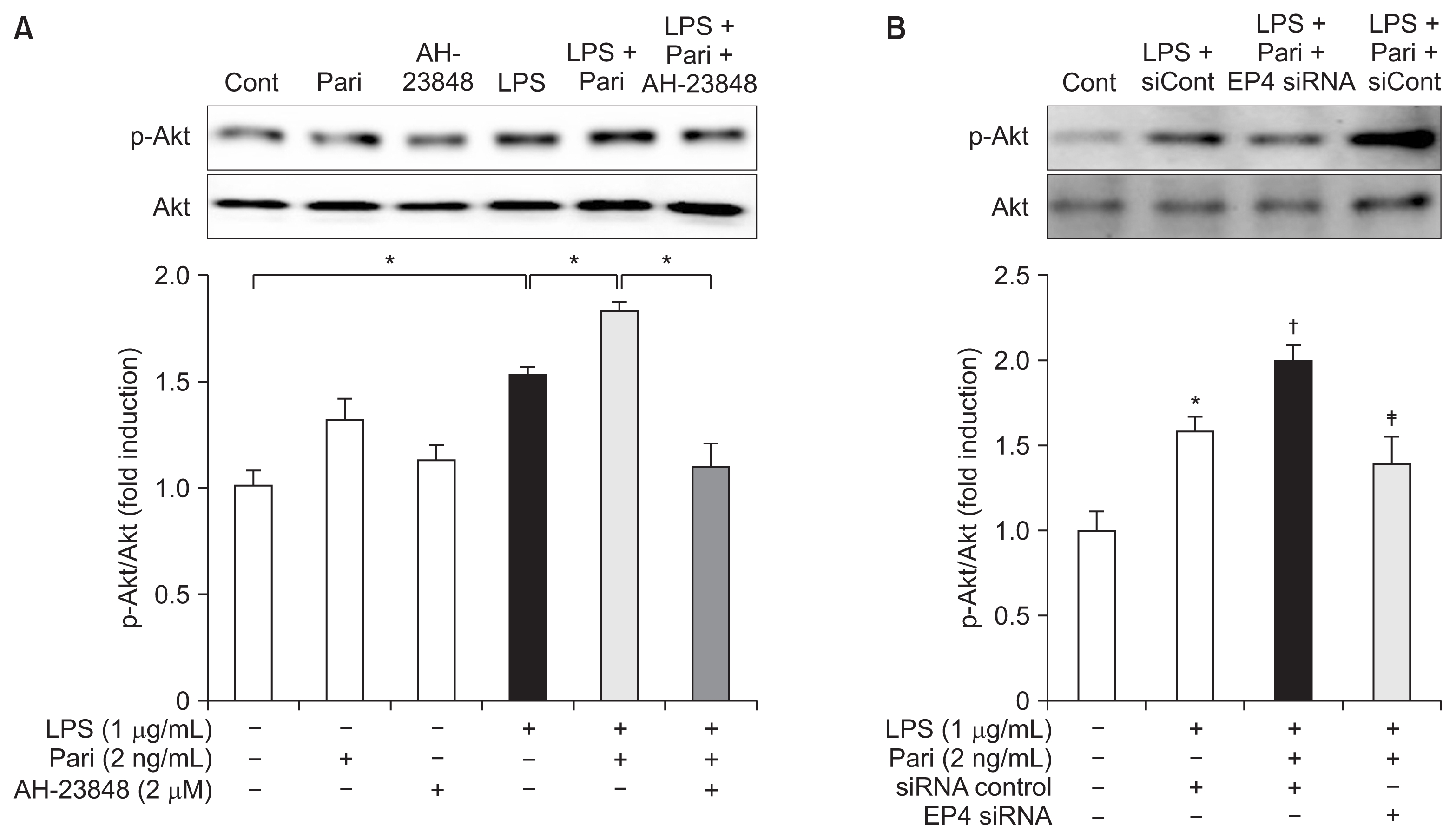
Paricalcitol induces EP4-mediated phosphorylation of Akt in HK-2 cells with endotoxin exposure
(A) Semiquantitative immunoblot analysis showed that endotoxin induced Akt phosphorylation and that it was further enhanced after paricalcitol. The upregulated Akt phosphorylation in paricalcitol-treated cells was attenuated by AH-23848 co-treatment. (B) EP4-specific siRNA significantly inhibited the phosphorylation of Akt in LPS-exposed cells treated with paricalcitol.
Cont, control; LPS, lipopolysaccharide; Pari, paricalcitol; siCont, control siRNA; siRNA, small interfering RNA.
Each column represents the mean ± SEM of three independent experiments. *P < 0.05 vs. Cont group; †P < 0.05 vs. LPS + siCont group; ‡P < 0.05 vs. LPS + Pari + siCont group.
The EP4-dependent pathway contributes to the antiapoptotic effects of paricalcitol in LPS-exposed HK-2 cells
To evaluate the effect of paricalcitol on LPS-induced apoptosis, HK-2 cells stained with a TUNEL assay were analyzed after treatment with LPS with or without paricalcitol. The degree of apoptosis was expressed as the number of TUNEL-positive cells (Fig. 7). LPS caused an increase in the number of apoptotic cells. Paricalcitol significantly decreased the number of TUNEL-positive cells, whereas EP4 siRNA increased the number of TUNEL-positive cells compared to that of LPS-exposed cells treated with paricalcitol alone.
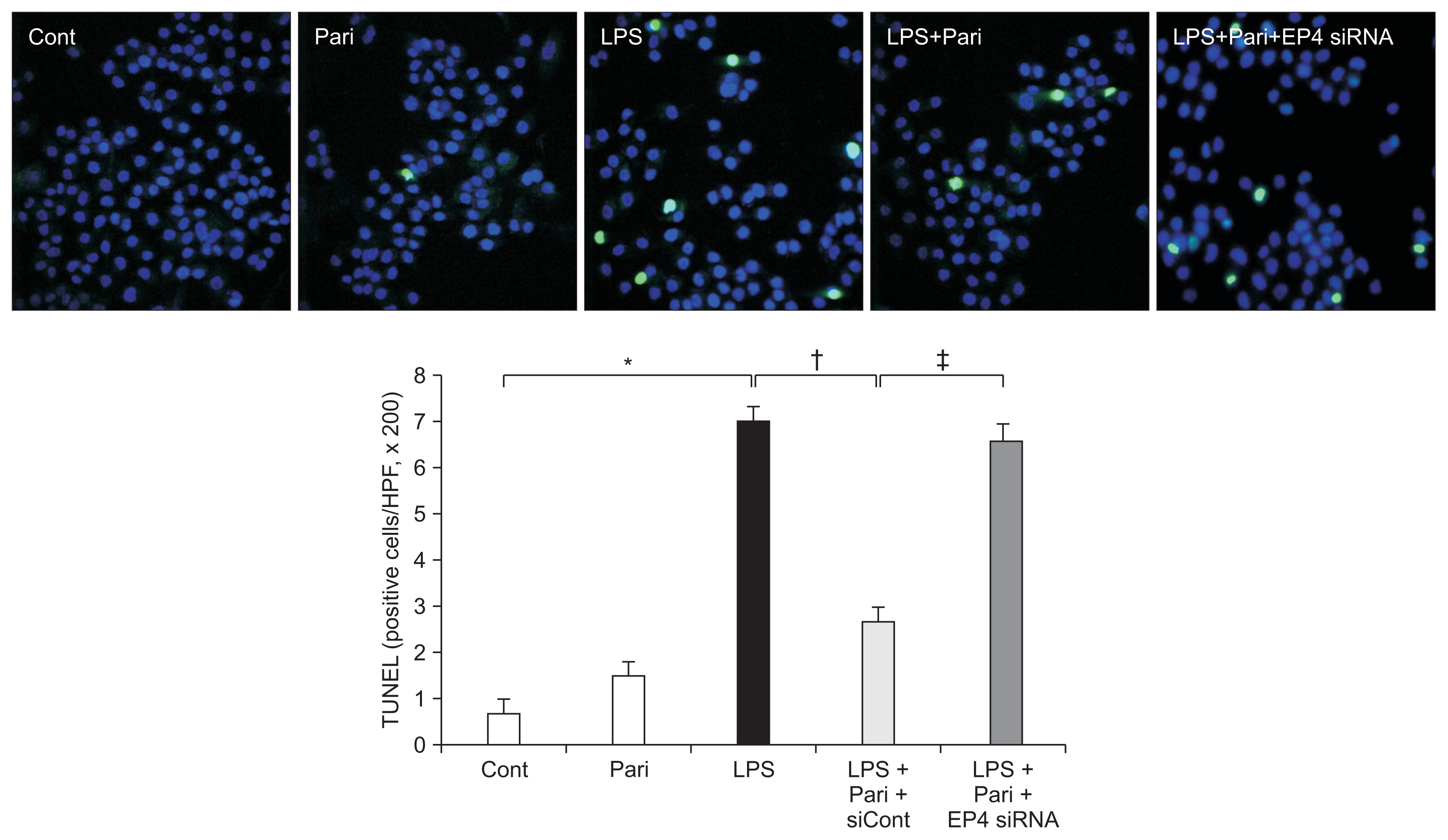
Paricalcitol attenuates apoptosis via an EP4-dependent pathway in HK-2 cells with endotoxin exposure
TUNEL assay revealed that paricalcitol pretreatment significantly reduced the number of TUNEL-positive cells in LPS-exposed HK-2 cells, whereas the number of TUNEL-positive cells was increased by the treatment of EP4-specific siRNA. Original magnification × 400.
Cont, control; LPS, lipopolysaccharide; Pari, paricalcitol; siRNA, small interfering RNA; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling.
*P < 0.05 vs. Cont group; †P < 0.05 vs. LPS; ‡P < 0.05 vs. LPS + Pari.
Discussion
This study demonstrated that paricalcitol pretreatment had a protective effect against endotoxin damage in proximal tubular cells through the PGE2–EP4-signaling pathway. The paricalcitol upregulated the cellular membrane expression of EP4 and increased PGE2–EP4 binding. This EP4 activation attenuated HK-2 cell death on LPS exposure and induced the changes in its downstream signaling pathway including the phosphorylation of Akt and inactivation of p65 NF-κB and reduced several inflammatory cytokines and cell apoptosis. The protective effects of paricalcitol and changes in the EP4 downstream signal were definitely abolished with EP4 antagonist treatment or EP4–siRNA transfection. These findings suggest that paricalcitol improved inflammation and apoptosis of proximal tubular cells through activation of EP4 and its downstream signaling pathway.
LPS is a potent stimulant of inflammatory cell activation and evokes the secretion of proinflammatory mediators. PGE2 is one of the representative mediators that are increased in immune cells and glomerular mesangial cells after LPS exposure and COX-2 is a major synthetic enzyme of PGE2 [13,14]. In the present study, LPS stimulation increased the expression of COX-2 and PGE2 production in HK-2 cells and paricalcitol pretreatment further increased their expression. We also found that paricalcitol treatment enhanced the cellular membrane expression of EP4 in LPS-exposed cells and that binding between PGE2 and EP4 was enhanced in LPS-exposed paricalcitol-treated cells. These findings suggest that the responses of COX-2–PGE2 pathway to LPS treatment in HK-2 cells are similar to in vitro studies using different kinds of cell type and that paricalcitol reinforces the up-regulation of COX-2 and PGE2 signaling and their biological action.
In this study, we found that paricalcitol treatment increased the expression of EP4 mRNA, but the mRNA expression of EP1–EP3 was not changed. These findings suggest that paricalcitol upregulates the PGE2 receptor EP4 more selectively. It was recently established that EP4 stimulation can activate the PI3K/Akt pathway, which results in the transcriptional activation of T-cell factor [15]. This activation increases the expression of COX-2, which leads to greater synthesis of PGE2. As a result, EP4 stimulation has the potential to set a positive feedback loop that would further stimulate its own receptor [16,17]. Therefore, we suggest that a positive feedback between COX-2 and EP4 might involve the EP4 selectivity of paricalcitol and that increased phosphorylation of Akt and greater interaction between PGE2 and EP4 support the concept of a positive feedback loop.
NF-κB is one of the important transcription factors initiating the inflammatory cascade and regulating the gene expression of cytokines, chemokines, and adhesion molecules [18]. Previous studies using various models of renal disease suggested that paricalcitol has inhibitory effects on NF-κB [7,8,19,20]. However, there were no previous studies to elucidate how paricalcitol regulates the NF-κB activity to prevent proximal tubule cell injury, especially in a model of sepsis-induced AKI. In the present study, we set inflammatory conditions in HK-2 cells using LPS stimulation, and our data indicated that paricalcitol pretreatment attenuates LPS-induced cell death that depends on the EP4 pathway and that EP4 controlled the inhibitory effect of paricalcitol on NF-κB phosphorylation and nuclear translocation. To determine the functional role of EP4 with paricalcitol effects further, EP4 siRNA was used to investigate the effect on TNF-α, RANTES, MCP-1 and IL-1β, which was transcriptionally activated by NF-κB. As expected, paricalcitol obviously suppressed these proinflammatory cytokines in an EP4-dependent manner. These findings suggest that the EP4-dependent pathway is one of the major anti-inflammatory mechanisms of paricalcitol in endotoxin-induced tubular cell injury.
PI3K is a main target of PGE2–EP4 signaling, and its activation subsequently increases the phosphorylation of Akt kinase [11]. The phosphorylation of Akt is a representative signal that leads to antiapoptotic effects and cell survival via upregulation of several downstream kinases [12,21,22]. In the present study, paricalcitol increased the phosphorylation of Akt in LPS-treated cells and an EP4 blockade reversed the phosphorylation of Akt. In addition, a TUNEL assay showed that apoptotic cell death from LPS treatment was significantly attenuated in paricalcitol-treated cells via the EP4-dependent pathway. These findings suggest that the antiapoptotic effect through an EP4-specific pathway is a critical protective mechanism of paricalcitol in LPS-exposed HK-2 cells.
Paricalcitol significantly increased VDR expression in the present study. It seems reasonable, but raises the interesting questions why paricalcitol, VDR activator, increases the amount of VDR protein and whether beneficial effects of EP4 activation is associated VDR upregulation. While this experiment did not show clear mechanism on this result, we presume that paricalcital may activate the positive loop, which involves the increment in protein level of VDR [23]. Moreover, paricalcitol-induced caspase inhibition may increase the VDR expression, because the activation of caspase-3 enhances VDR cleavages [24,25]. In addition, we suggest that VDR upregulation potentially mediated the EP4 activation. The EP4 promoter includes nucleotide sequences of vitamin D response element, which potentially binds VDR and activates EP4 signaling pathway [26–28].
Vitamin D deficiency has a close relationship to the adverse outcomes of sepsis, including increased length of hospitalization, sepsis-induced AKI, and mortality [29–31]. Paricalcitol has been widely used in various diseases with few adverse reactions and good tolerance. Our study suggests that paricalcitol may have potential as a therapeutic intervention for septic AKI via the EP4-dependent pathway. However, the limitation of our experimental design was that paricalcitol was applied prior to the LPS injury and there is insufficient in vivo evidence. In addition, this study also had a limitation to demonstrate the EP4 own role in LPS-injured cells. Further in vivo experiments and clinical studies to investigate the therapeutic role of EP4 activation are needed.
In conclusion, our study suggested that paricalcitol up-regulated EP4 expression in HK-2 cells after LPS injury, and the subsequent activation of the EP4 pathway mediated activation of Akt and inhibition of NF-κB. These actions of paricalcitol on PGE2–EP4-signaling pathway contributed to its anti-inflammatory and antiapoptotic properties in LPS injury. Therefore, paricalcitol treatment provides a new therapeutic strategy for sepsis-induced AKI through activation of PGE2–EP4 receptor signaling.
Supplementary Data
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2015R1A1A1A05001599), and Clinical Research Institute grant (CMCDJ-A-2015-007) funded by The Catholic University of Korea Daejeon St. Mary’s Hospital. The authors also wish to acknowledge the financial support of the Korean Society Nephrology (Chong Kun Dang, 2016).
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.