


Introduction
This discussion focuses on structural features of diabetic nephropathy (DN), and how these can be used to point to potential clinical interventions that may ameliorate and potentially reverse established DN. The features of DN have been well established [1]. There is a well described sequence of glomerular injury beginning with hypertrophy of the glomerulus. Hypertrophy refers to an enlargement of all the components of the glomerulus, which is thought to be a consequence of the physiologic changes of hyperfiltration that occur early in the course of diabetic renal injury. Subsequent morphologic changes within the glomerulus traditionally have focused on the mesangium, because it is the most histologically recognized feature of DN and the feature most easy to quantify, both in renal biopsies obtained from patients with DN and in animal models used to study DN. However, the mesangial expansion, no matter how prominent, is unlikely to perturb the glomerular filtration barrier in a way that is sufficient to cause the marked proteinuria, often in nephrotic range, that is a common feature of DN. In thinking about the glomerular injuries of diabetes, we want to be inclusive of changes in the glomerular filtration barrier, including podocytes and the endothelium, and consider how these may interact with the mesangial changes that are so prominent and characteristic in advanced DN to produce the structural and functional injuries of DN.
Our understanding of these changes has been greatly informed by extensive studies of sequential protocol biopsies obtained from type I diabetic patients by Fioretto et al [2], and by biopsies obtained from the Pima Indian population of the southwestern United States, which has a high prevalence of type II diabetes and DN [3].
Sequence of histologically recognizable changes in progressive diabetic nephropathy
The development of human DN is a complex process that involves interactions of all of the cells which comprise the glomerular tuft. The morphologic alterations of diabetic glomerular injury begin as a process of hypertrophy. The structural relationships of the glomerulus remain unchanged, but the glomeruli, and indeed the entire renal parenchyma, enlarges as a structural response to the hyperfiltration that is a physiologic feature of early diabetic renal injury [1].
Development of the most pathognomonic lesion of morphologically advanced DN, that of diffuse mesangial sclerosis, in both type I and type II diabetics, a characteristic sequence of progressive mesangial alterations can be identified. However, over a period of time, typically years, following the development of hypertrophy the mesangium becomes expanded due to accumulation of extracellular matrix. This process is progressive and the accumulation of matrix can lead to a nodular configuration of the expanded mesangium. Critical to our understanding of this process of mesangial expansion is that we recognize that this expansion consists not only of a linear process of continuous matrix accumulation, but also involves processes of active injury (mesangiolysis) and repair. Mesangiolysis refers to the dissolution of the mesangial architecture and disruption of the matrix comprising the mesangial nodules, and/or fraying of the interface between the mesangium and the adjacent glomerular capillaries. Analysis of the pathology of the characteristic nodules, often referred to as Kimmelstiel-Wilson nodules, using special histology stains such as silver methenamine, delineates this process of repetitive mesangiolysis and repair. The evidence of repetitive injury comes from the laminated appearance of the mesangial matrix revealed with a silver stain; the evidence of repair comes from demonstration of vascular channels (neo-angiogenesis) that is often identified in portions of the expanded mesangial matrix [4]. The evidence that this is an active process of repetitive injury and repair points to the possibility of therapeutic interventions that either abrogate the specific active injury of mesangiolysis or promote an ongoing reparative process. To date, we lack sufficient molecular characterization of the biologic pathways involved in this specific injury to identify specific peptides or gene targets that can be therapeutically manipulated to enhance the reparative process in this location and in this setting. This can be addressed by better characterization and wider use of appropriate model systems that would support preclinical investigation and identification of such pathways, and by testing interventions in preclinical models of diabetic glomerular injury that specifically focus on the processes of mesangiolysis and repair.
Podocytes are essential for the maintenance of mesangial integrity. Although we lack precise biochemical pathways that mediate the process of mesangiolysis, there is evidence that comes from both animal models and from human biopsies that implicate the loss of podocytes, or loss of key physiologic functions of podocytes, in the development of mesangiolysis. A relevant animal model is a unique transgenic mouse created by Matsusaka et al [5], who placed CD25, the receptor for the immunotoxin LMB2, on podocytes. When LMB2 is administered to mice, there is widespread podocyte injury and death. This model was used initially to examine the role of podocyte number and density in the development of lesions of focal and segmental glomerulosclerosis, but it was apparent in the first descriptions of the model that widespread loss of podocytes also led to a process of mesangiolysis. A second line of evidence that loss of podocyte function can lead to mesangiolysis comes from the extensive studies by Eremina et al [6] and Sison et al [7] which have used genetically manipulated mice to diminish or knockout the production of vascular endothelial growth factor (VEGF) in podocytes. While complete knockout of VEGF is lethal in developing mice, a 75% reduction in gene dose can lead to survival but early mortality of young mice. This diminished production of VEGF by podocytes is accompanied by dramatic lesions of mesangiolysis. The implication of these studies is that podocyte production of VEGF provides a trophic function, whether directly or indirectly, for mesangial cells and is important for the maintenance of mesangial integrity. This is further confirmed by studies in human patients exposed to the VEGF antagonist, bevacizumab, a therapeutic agent utilized to treat cancers by blocking tumor angiogenesis. An important side effect of administration of this drug is the development of hypertension and proteinuria in a significant number of patients, and the development of mesangiolysis and a thrombotic microangiopathy in a small number of severely affected patients [8]. Taken together, it appears there is an essential role in both mice and humans for podocyte function, at least in part through the production of VEGF, to preserve mesangial integrity.
A critical finding of the prospective studies of kidney biopsies from type I diabetics and of Pima Indians is that total podocyte number was diminished in both populations as diabetic glomerular injury progressed. These findings suggest that with progressive loss of podocytes, there is a complex interaction in the glomerulus that links the progressive loss of podocytes to repetitive injuries of mesangiolysis and to the eventual development of the nodular glomerulosclerosis that is such a characteristic lesion of morphologically advanced DN. The mechanism by which podocyte production of VEGF directly affects the mesangium remains poorly delineated. There is evidence from murine systems that VEGF binds to its receptors flt and flk on the endothelium that lines the glomerulus. Engagement of these receptors by the VEGF ligand is thought to be important for maintaining the characteristic fenestrated appearance of this endothelium. It is reasonable to speculate that key trophic factors that support the maintenance of mesangial integrity are derived from the glomerular endothelium, which is in direct contact with mesangial structures. In such a scenario, the endothelial factors responsible for this process are first supported by the trophic functions of VEGF acting directly on the endothelium, which then produces other, as yet unspecified, factors to support the mesangium. The details of this postulated relationship between podocytes, endothelial cells, and the mesangium in diabetic renal injury remains largely obscure. However, there has been a recent focus on the endothelium in diabetic renal injury, and in particular on the renal glycocalyx which sits at the endothelial surface facing the lumen of glomerular capillaries and which is exposed to the capillary circulation. The endothelial glycocalyx is composed of a complex mixture of proteins and proteoglycans, and these protein and proteoglycans entities are capable of binding specific cytokines and growth factors. It has been shown in recent studies by Boels et al [9,10] that this glycocalyx is diminished in experimental models of DN, and that some therapeutic interventions can serve to modulate the endothelial glycocalyx and restore it to at least normal size using some of the therapeutic interventions currently being tested to ameliorate DN. These topics—the ability of podocytes to support endothelial integrity; the maintenance of a normal glycocalyx; the characterization of specific composition of the glycocalyx as it is modified in disease settings and the consequences for binding of specific cytokines and trophic growth factors; and the relationship of these systems to the maintenance of the integrity of the mesangium are all avenues deserving of further exploration as we seek to identify new and better therapeutics for patients with diabetic kidney disease.
Our studies of the BTBR ob/ob mouse demonstrated that it is a good model of DN and type II diabetes [11]. These mice develop manifestations of morphologically advanced DN including progressive mesangial expansion due to increased matrix, mesangiolysis, basement membrane thickening, and marked proteinuria [12]. This injury is a function both of the unique insulin resistance of the wild type BTBR mouse, and the introduction of the ob/ob mutation which results in leptin deficiency. With leptin deficiency, the mice become hyperphagic, obese, and develop further insulin resistance, and diabetes. They appear uniquely susceptible to a number of diabetic complications including nephropathy, neuropathy, and cardiomyopathy. A dramatic finding of these mice is that these morphologic changes and functional changes of advanced DN can be reversed by the administration of leptin, which abrogates the hyperphagy of these mice and results in weight loss and restoration of normoglycemia [13].
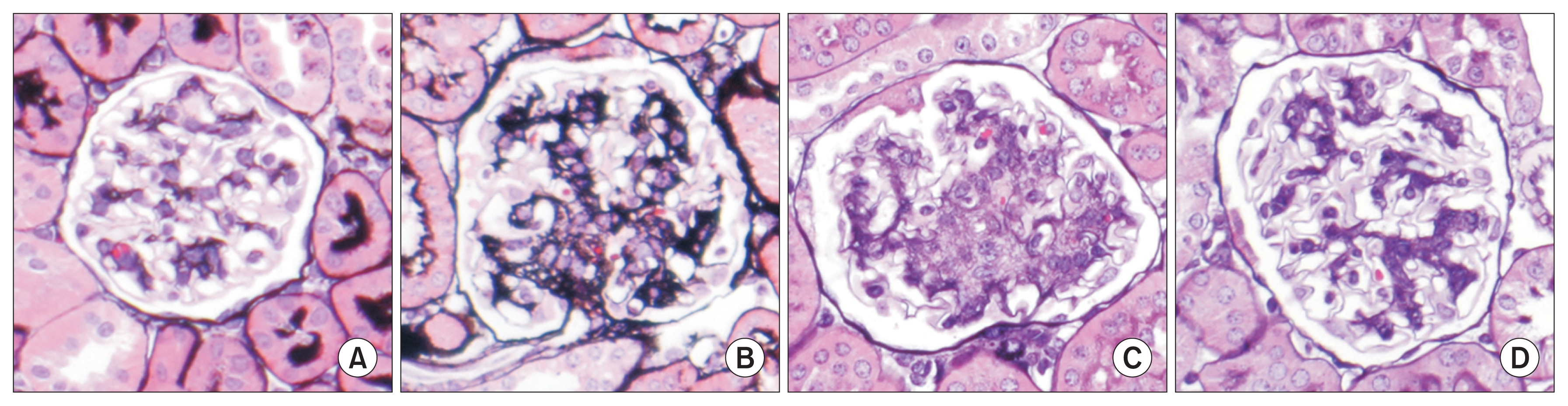
As has been previously reviewed [11], this model has some advantages over some other widely used murine models of DN by the more extensive accumulations of mesangial matrix resembling more advanced DN as it occurs in humans, especially when compared with such widely used murine models as Akita mice and streptozotocin induced diabetes in C57BL/6 mice (both models of type I diabetes), and db/db mice with deficiency of the leptin receptor (a widely used model of type II diabetes). These mice develop more modest mesangial expansion, and hence at best model earlier stages of glomerular injury due to diabetes. The BTBR ob/ob mice develop lesions more quickly than these other mice, which increases their suitability for intervention studies. Most importantly, this is the only murine model which has demonstrable reversibility of lesions, which allows for studies of mechanisms underlying this process that are not possible with other current leading murine models (Fig. 1). Disadvantages of the BTBR ob/ob model include the susceptibility of these mice to early mortality by the age of 24 weeks, their propensity to develop infections (likely a function of both their obesity and diabetes, which also reflects a common phenotypic feature of human diabetic complications), and the infertility of mice homozygous for the ob/ob mutation.
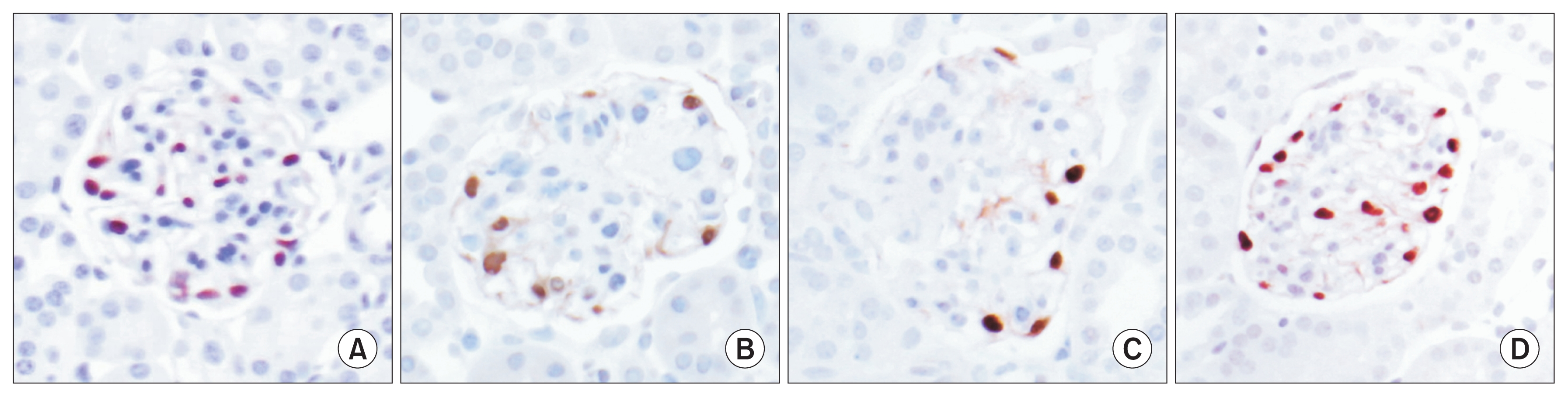
As we have previously reported, the restoration of normal glomerular structure following six weeks of administration of leptin via minipump infusion also results in restoration of normal podocyte number and density (Fig. 2). Treatment of mice for a similar period of time using the leading therapeutic agents currently employed to ameliorate DN, the angiotensin converting enzyme enalapril and the angiotensin receptor blocker losartan, failed to reverse the DN encountered in these mice and, in particular, failed to restore normal podocyte number [13]. We have shown that one potential source of the restored podocytes may derive from a progenitor cell population within the parietal epithelial cell (PEC) population.
Lessons from the BTBR ob/ob model of DN may be particularly useful in strategizing for the development of new therapeutics. In this model, the development of DN and the characteristic mesangial injuries are preceded by the loss of podocytes. Podocytes are lost by eight weeks of age in this model, and are associated with the onset of proteinuria at about this time. Histologically detectable expansion of the mesangium occurs by the age of 10 weeks and is then progressive. We have shown that replacement of the key molecule, leptin, which is not produced by virtue of the ob/ob mutation, leads to restoration of podocytes, while other conventional therapies utilized for DN, such as renin-angiotensin inhibition do not. Therefore, this model may be particularly useful for testing the efficacy of therapeutics directed towards podocyte restoration. We have suggested, based on the finding that enalapril and losartan administered in a six-week therapeutic protocol comparable to that utilized for leptin replacement, do not lead to comparable restoration of podocytes and that this may be a limitation of the efficacy of these agents in human DN, where they slow but do not reverse progress of the disease.
The studies raise the important question of what may be the source of restored podocytes in reversible DN. It has long been established that podocytes are terminally differentiated cells with limited replicative capacities. Romagnani and others have provided evidence for a potential progenitor cell population within the glomerular PEC layer that may be a potential source for new podocytes [14,15]. Our study supports a hypothesized scenario whereby podocyte restoration is enabled by mobilization of an activated PEC population.
Others have suggested such scenarios of podocyte regeneration from PEC precursors are unlikely, based on studies of mature strains of mice other than BTBR [16]. This issue of whether podocytes can be replenished from a PEC precursor population remains controversial and requires greater investigation. Other possible sources of regenerating podocytes include cells of renin lineage located in the juxtaglomerular apparatus, as revealed by studies from Eng et al [17] and Pippin et al [18,19]. However, the idea that podocyte regeneration can occur, and that DN can be reversed, holds great promise for the development of improved therapeutics for DN. In a landmark study by Fioretto et al [20], it was demonstrated that pancreatic transplantation in diabetic patients with biopsy proven, morphologically advanced DN who have had successful function of the pancreatic transplants for a period of 10 years or more can have reversal of the lesions of advanced DN. Providing support for the concept of podocyte restoration as a key component of this scenario in humans, we have recently shown that there may be a balance between podocyte loss and podocyte restoration in DN in humans, particularly in the early stages of the evolution of the glomerular injury [21]. This proof of principle that reversal can occur in both humans as well as in mouse models should provide us with the incentive to continue to focus on a cure, and not just amelioration, of DN.
An important caveat to this review is that we have focused exclusively on diabetic injuries to the glomerulus that are largely a consequence of loss of podocyte function. We have not considered DN as a tubulointerstitial or vascular disorder. We also have not considered such important issues as inflammation or hemodynamics or hypertension in contributing to the structural and functional manifestations of advanced DN. Modification of each of these, in turn, may also contribute to the ultimate goal of reversal of DN.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









