Dihydroxyadenine crystal-induced nephropathy presenting with rapidly progressive renal failure
Article information
Abstract
Adenine phosphoribosyltransferase enzyme deficiency is a rare, autosomal recessive disorder. It is a disease limited to the renal system and usually presents with urolithiasis. Herein, we report a young female with dihydroxyadenine (DHA) crystal-induced nephropathy presenting with rapidly progressive renal failure. DHA crystals can be easily diagnosed by their pathognomic color and shape in urine and biopsy specimens. A high index of clinical suspicion helps in the early diagnosis of this potentially treatable renal disease.
Introduction
Adenine phosphoribosyltransferase (APRT) deficiency is an uncommon, autosomal recessive disorder of purine metabolism [1]. The most common clinical manifestation of APRT deficiency is formation of a kidney stone. APRT deficiency can also produce crystal nephropathy that may lead to chronic kidney disease (CKD). However, a significant number of patients remain asymptomatic. There is no specific manifestation of this disease; therefore, awareness of APRT deficiency is important to diagnose this condition. We present a patient with rapidly progressive renal failure secondary to dihydroxyadenine (DHA) crystal-induced nephropathy that was confirmed by kidney biopsy. Timely diagnosis and treatment resulted in rapid improvement of the patient’s renal function.
Case report
A 24-year-old female born to a consanguineously married couple presented with complaints of nausea, vomiting, anorexia, and fatigue for three months. There was no history of pedal edema, decreased urine output, or intake of any nephrotoxic drugs. The patient denied a family history of similar illness.
Her clinical examination was unremarkable. Her laboratory investigations revealed serum creatinine of 1.86 mg/dL, which further increased to 2.4 mg/dL within four days of admission. Serum complement levels were within normal range. Her serum calcium was 11.2 mg/dL, 25-hydroxyvitamin D was 175 nmol/L (50–250 nmol/L), and parathormone was 1.28 pmol/L (1.3–6.8 pmol/L). Serum sodium was 137 mmol/L, and serum potassium was 4.16 mmol/L.
Her blood count revealed hemoglobin 10.7 g/dL, total leucocyte count 8,900 × 109/L, and platelet count 279 × 109/L. Renal ultrasound revealed normal sized kidneys with no renal stones. Urinalysis showed 7.2 pH, no red blood cells, and 10 to 15 white blood cells/high power field. Urine culture showed no growth.
With a provisional diagnosis of tubulointerstitial disease, a renal biopsy was performed and revealed two cores of renal parenchyma with 25 unremarkable glomeruli with predominantly tubulointerstitial injury. The tubular lumen revealed numerous brown-green crystals of varying shapes (rod, rhomboid, granular, fan shaped or radially arranged), eliciting foreign body reactions (Fig. 1A). These crystals were also seen in the cytoplasm of tubular epithelial cells, interstitium, blood vessels, and macrophages (Fig. 1B, C). These crystals were weak periodic acid–Schiff positive, argyrophilic with silver methenamine, and bluish green on Masson’s trichrome stain (Fig. 1D, E). The crystals were birefringent under polarizing microscopy (Fig. 1F). The crystals showed a characteristic brownish color, and DHA crystal-induced nephropathy was diagnosed.

Renal biopsy findings
(A) Marked tubular atrophy, interstitial inflammation, and fibrosis are shown. Higher magnification shows 2,8-dihydroxyadenine (DHA) crystals within the tubular lumens (B) with giant cell reaction in the interstitium and also in the vascular lumen (C). (D, E) The crystals are argyrophilic with silver methenamine and bluish green on Masson’s trichrome stain. (F) The crystals are seen within a tubular lumen under polarized light microscopy.
A repeat urine sample was analyzed, and light microscopy of the urine sediment showed 2,8-DHA crystals. Polarization of these crystals revealed the characteristic Maltese cross pattern (Fig. 2). The patient was started on allopurinol and prescribed a low salt, low fat, and high fluid diet. Her constitutional symptoms rapidly improved. The patient was asymptomatic with normal renal function at the six month follow-up visit.
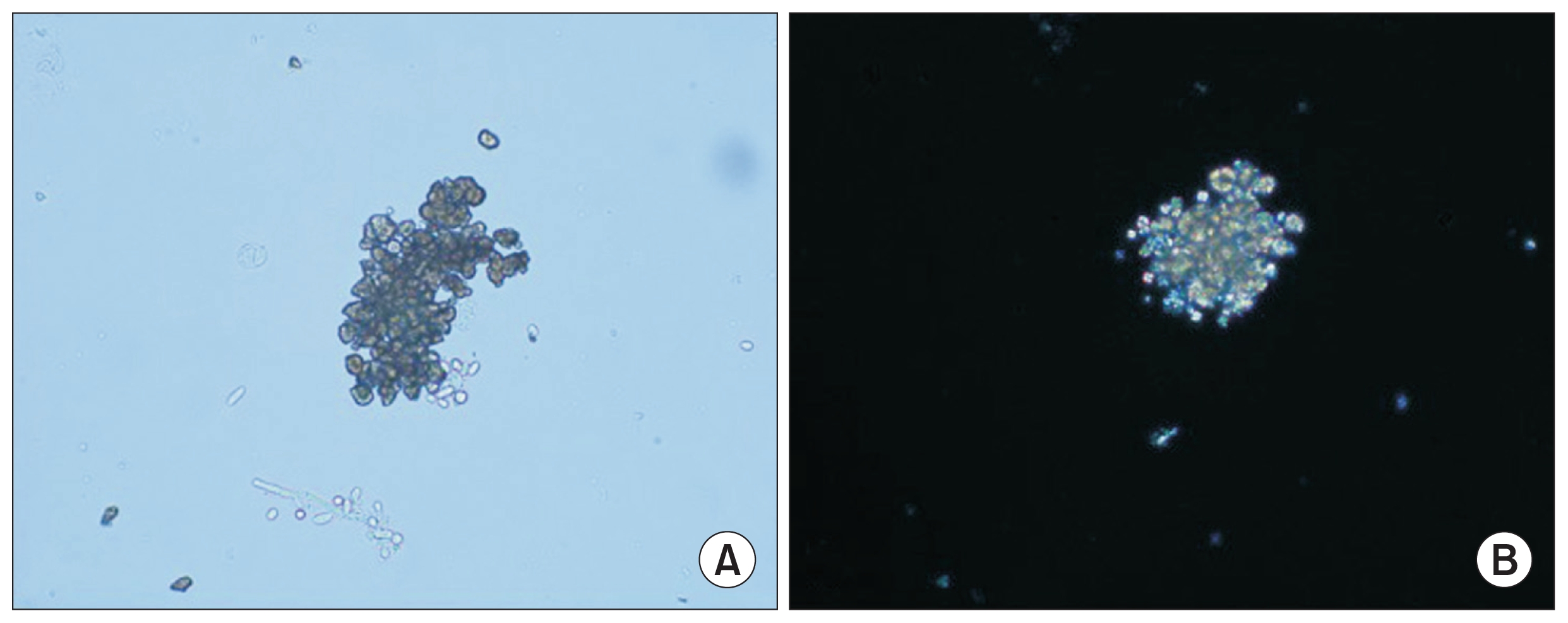
Urine examination (×40)
(A) The typical brown dihydroxyadenine crystals have a dark outline and central spicules in conventional light microscopy. (B) The crystals appear yellow under polarized light microscopy.
Discussion
The APRT enzyme is necessary for the metabolism of adenine to adenosine monophosphate in the presence of phosphoribosyl pyrophosphate. With deficiency of the APRT enzyme, xanthine dehydrogenase (XDH) converts adenine to 2,8 DHA, which is excreted by the kidneys. DHA is protein bound in plasma, but it is insoluble in urine at any physiological pH, which leads to crystalluria and, less commonly, crystalline nephropathy (Fig. 3).

Mechanisms of adenine phosphoribosyltransferase (APRT) deficiency
Adenine cannot be converted to adenosine and is thus oxidized to 2,8 dihydroxyadenine (DHA) by xanthine dehydrogenase (XDH) via an 8-hydroxy intermediate.
AMP, adenosine monophosphate.
Nearly 10% of patients with DHA crystaluria progress toward end stage renal disease and may require renal replacement therapy [2]. In many cases, the diagnosis of APRT deficiency was first recognized after dysfunction of a transplanted kidney [3,4]. At least 50% of patients with DHA crystaluria are asymptomatic until adulthood, and their CKD is usually attributed to obstructive uropathy and nephrolithiasis-related chronic tubulointerstitial nephritis. The exact cause of the phenotypic variability is not known; however, hydration status and excess intake of purine-rich food can affect presentation [5,6]. In our case, hypercalcemia could have acted as a triggering factor for crystallization. Although the etiology of hypercalcemia in our patient could not be ascertained, the treating team thought it could be due to consumption of calcium and vitamin D for her spondylolisthesis. Because the patient improved after initiation of treatment, a second kidney biopsy was not ethical, and the hypothesis of hypercalcemia triggering crystallization could not be confirmed.
Diagnosis of APRT deficiency is suggested by 1) detection of urinary 2,8-DHA crystals, 2) histologic findings of crystal nephropathy, and 3) kidney stone analysis. The disease can be confirmed by 1) genetic testing and 2) demonstration of absence to APRT activity in red blood cell lysate.
APRT deficiency is a rare disease. However, misidentification of 2,8-DHA crystals (radiolucent kidney stones) as uric acid calculi or xanthine stones is a significant cause of delayed diagnosis. We summarize the differences in the appearance of DHA crystals, uric acid stones, oxalate, and xanthine stones in Table 1.

Salient features of the differential diagnosis in patients with metabolic crystalline nephropathy
Since DHA stones are radiolucent, they are usually first diagnosed on ultrasonography or computed tomography scan either incidentally or during work up for nephropathy. A careful urinary examination is usually sufficient to diagnose DHA stones. However, the diagnosis can be confirmed by biochemical stone analysis, APRT enzyme assays from erythrocyte lysate, and genetic analysis by sequencing, which has shown more than 40 mutations of the APRT gene.
Renal biopsy plays a very important role in patients with renal dysfunction in the absence of renal stones. The renal histopathology in patients with APRT deficiency is characterized by diffuse DHA crystal deposition and tubulointerstitial abnormalities. As with urine microscopy, the DHA crystal appearance may be differentiated from hyperoxaluria and hyperuricemia (Table 1). It has been studied that evaluation of crystals in urine samples/renal biopsy by Fourier transform infrared microscopy are highly specific, and measurement of APRT activity level in erythrocyte lysate should also be done for final confirmation of the diagnosis. Genetic testing may be done, but it is not necessary for diagnosis. These diagnostic modalities are not freely available (especially in underdeveloped countries), and a careful urine examination and kidney biopsy evaluation are often sufficient for reaching the diagnosis.
We reviewed other cases of DHA crystal nephropathy in the English published literature (Table 2). To the best of our knowledge, ours is the third case reported from India [7,8]. However, the patient in our case did not have a history of renal stones preceding development of renal failure.

Comparison of the clinicopathological features and treatments in Indian studies
Timely recognition of the crystals in urine or on biopsy is important because initiating a purine-poor diet can retard the formation of crystals, and strong diuresis can wash out already-formed crystals. Recently, the nonpurine XDH inhibitor febuxostat has been used as an alternative therapeutic option. Edvardsson et al [9] compared the effects of both drugs on level of urinary 2,8 DHA excretion in a small cohort of patients and found significant reduction in DHA excretion with febuxostat.
In conclusion, DHA crystals can be easily diagnosed by their pathognomic color and shape in urine and biopsy specimens. Due consideration should be given to a young patient presenting with acute or CKD with nephropathy and crystals in the urine without urolithiasis. A high index of clinical suspicion helps in the early diagnosis of this potentially treatable renal disease.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.