Renal sympathetic nerve activation via α2-adrenergic receptors in chronic kidney disease progression
Article information
Abstract
Chronic kidney disease (CKD) is increasing worldwide without an effective therapeutic strategy. Sympathetic nerve activation is implicated in CKD progression, as well as cardiovascular dysfunction. Renal denervation is beneficial for controlling blood pressure (BP) and improving renal function through reduction of sympathetic nerve activity in patients with resistant hypertension and CKD. Sympathetic neurotransmitter norepinephrine (NE) via adrenergic receptor (AR) signaling has been implicated in tissue homeostasis and various disease progressions, including CKD. Increased plasma NE level is a predictor of survival and the incidence of cardiovascular events in patients with end-stage renal disease, as well as future renal injury in subjects with normal BP and renal function. Our recent data demonstrate that NE derived from renal nerves causes renal inflammation and fibrosis progression through alpha-2 adrenergic receptors (α2-AR) in renal fibrosis models independent of BP. Sympathetic nerve activation-associated molecular mechanisms and signals seem to be critical for the development and progression of CKD, but the exact role of sympathetic nerve activation in CKD progression remains undefined. This review explores the current knowledge of NE-α2-AR signaling in renal diseases and offers prospective views on developing therapeutic strategies targeting NE-AR signaling in CKD progression.
Introduction
Chronic kidney disease (CKD) progression, ultimately leading to end-stage renal disease (ESRD) remains a significant health burden with its pathogenesis poorly defined. Along with an increase in metabolic disease, hypertension, obesity, diabetes and aging, the prevalence of CKD has grown in developed countries [1]. Patients who have severe CKD or ESRD are exposed to the high risk complicated outcomes of cardiovascular disease, stroke, and death [2]. Furthermore, recent reports demonstrate that acute kidney injury (AKI) is a significant risk for onset or acceleration of CKD [3,4].
The sympathetic nervous system controls the physiological functions of diverse organ systems, including the kidney [5]. The catecholamines epinephrine (adrenaline), synthesized in chromaffin cells of the adrenal medulla, and norepinephrine (NE; noradrenaline), released by sympathetic neurons, are sympathetic nervous system effectors [6]. Epinephrine and NE act by binding to adrenergic receptors (AR), α1-AR, α2-AR or β2-AR, further classified by their subtypes, α1A-AR, α1B-AR, α1D-AR, α2A-AR, α2B-AR, α2C-AR, β1-AR, β2-AR, and β3-AR [6,7]. The use of mouse models with targeted deletions or transgenic over-expression of the respective genes in vivo has enabled the unraveling of the physiological and pharmacological functions of these individual receptor subtypes [8–11].
The kidney is innervated by efferent sympathetic nerves as well as peptidergic sensory afferent nerves [12,13]. Sympathetic nerve activity and the tissue content of neurotransmitters including NE is elevated in both patients and experimental animals with CKD [14–16]. Despite the recognition of the renal nerve as an effector of renal dysfunction in CKD [15,17,18], its role in the development and progression of CKD is not fully understood.
Renal denervation is a therapeutic strategy used in the treatment of resistant hypertension [19,20]. The beneficial effects of renal denervation against renal failure in both animals and humans include a decrease in BP, renal efferent sympathetic nerve activity, central sympathetic nerve activity and sympathetic outflow, and downregulation of the renin-angiotensin system (RAS), but the detailed molecular mechanisms remain elusive [13,21]. Several clinical trials in renal complications of hypertension and metabolic syndrome have been performed and are reviewed elsewhere [22–25]. Renal tubules as well as most inflammatory cells express ARs, including α2-AR. The presence of α2-AR in nephron segments, including proximal convoluted tubules and cortical and medullary collecting ducts, has previously been demonstrated [26]. We recently found that renal nerve-derived NE signaling via α2-ARs, α2A- and α2C-AR subtypes promotes renal inflammation and interstitial fibrosis in CKD disease progression models [27,28]. Here, we review the recent progress in our understanding of the molecular mechanisms of NE-AR signaling in renal disease development and progression.
Sympathetic nerve-derived norepinephrine is a profibrotic stimulator in injured kidneys
Regardless of the etiology of CKD, inflammation, and fibrogenesis are the common pathological processes that result in CKD and its progression to ESRD. We previously demonstrated that renal denervation can prevent fibrosis and inflammation in two different renal fibrosis models [27,28]. These results suggest that renal nerve stimulation may be a key mechanism driving renal inflammation and fibrogenesis, and that nerve-derived factors play a key role in the initiation of these processes.
NE, the primary neurotransmitter released by sympathetic nerve fibers, acts as a sympathetic activator in various bodily functions, causing increases in heart rate, arterial BP, tear production, and hepatic glucose production [29–32]. Furthermore, NE has both excitatory and inhibitory effects in various areas of the central nervous system [33]. In the kidney, NE can regulate renal blood flow, glomerular filtration rate, and tubular reabsorption of sodium and water, as well as release of renin and prostaglandins and neural control of renal function [13,19]. Our recent in vivo findings have shown that renal denervation in mouse kidneys prevents tubulointerstitial fibrogenesis after unilateral ureteral obstruction (UUO) and kidney ischemia/reperfusion injury (IRI) [27,28]. Interestingly, local infusion of NE into denervated kidneys increases transforming growth factor-β1 (TGF-β1) signaling, interstitial expression of α-smooth muscle actin (α-SMA), and excessive deposition of extracellular collagen matrix, mimicking the fibrotic response observed in the innervated kidneys [27,28]. As elevated plasma NE is observed in patients with CKD and ESRD [14,16], our study demonstrates that the IRI-induced increases in the level of NE may be a significant contributing factor to the development of IRI long-term sequelae in mice.
Norepinephrine is an inflammatory factor
The importance of inflammation in the development and progression of kidney fibrosis is well known. When kidney tissue is injured, inflammatory cells including lymphocytes, monocytes/macrophages, and dendritic cells infiltrate the site of injury and subsequently precede the process of kidney fibrosis through the release of fibrogenic cytokines and several growth factors [34]. The cytokines and growth factors activate fibroblasts and kidney tubular cells, which produce excessive extracellular matrix components at the injured site [34]. Monocytes/macrophages express most adrenoreceptor (AR) subtypes. Activation of α2-AR is responsible for upregulation of inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), while that of β2-AR confers an anti-inflammatory response [35]. NE regulates the production and secretion of TNF-α in macrophages [36–39]. NE affects myeloid cell recruitment into injured sites in sepsis models. Recent in vitro data show that NE regulates the cell fate and function of macrophages depending on the concentration of either endogenous NE or an AR agonist administered exogenously; a higher concentration of NE suppresses major histocompatability (MHC) class II and C-C chemokine receptor-2 (CCR2) expression and migration toward monocyte chemoat-tractant protein-1 (MCP-1), while a lower concentration enhances TNF-α expression and phagocytosis [35,40]. NE also promotes IL-12-mediated differentiation of CD4+ T cells into Th1 effector cells and subsequently increases the production of interferon-γ (IFN-γ) in the Th1 cells [41]. Conversely, NE reduces the production of Th1 cytokines, including IFN-γ and TNF-α, in hepatic T cells [42]. A recent report described the effect of NE on CD8+ T cell activity and differentiation via a β-AR-induced mechanism, and suggested that the time exposed to NE may influence the effect [35].
In contrast, studies on the effect of NE-AR on Treg and Th17 cells are very limited. A β2-AR agonist enhances the suppressive activity of Treg cells, leading to an increase in the anti-inflammatory response [43,44]. Furthermore, exposure of B cells to NE increases intracellular levels of IgG and IgE [45]. Studies investigating the effects of NE and β2-AR stimulation on B cell activity and T cell-dependent antibody response are reviewed elsewhere [46,47]. These studies indicate that, while the immune system is not absolutely dependent on the nerve system, NE can alter immune cell function resulting in either progression or protection against inflammatory diseases. Our recent studies using in vivo models of kidney interstitial fibrosis have shown that NE functions as a proinflammatory factor [27,28]. During kidney interstitial fibrogenesis after UUO and IRI, renal denervation suppresses the infiltration of polymorphonuclear (PMN)-positive neutrophils and F4/80-positive macrophages. Local infusion of NE into the denervated kidneys leads to the infiltration of neutrophils and macrophages, similar to the inflammatory response observed in the innervated kidneys [27,28], suggesting that NE is a key factor that induces inflammation after kidney injury.
Norepinephrine as a cell death inducer in injured kidneys
Tubular cell injury and death result in apoptotic bodies and other cellular debris, which are phagocytized by infiltrated macrophages, resulting in TGF-β1 release and extracellular matrix deposition [48]. The injured and disrupted kidney tubular cells also secrete proinflammatory cytokines such as IL-1, which upregulate adhesion molecules, including vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule-1 (ICAM-1), to facilitate the infiltration of inflammatory cells to the site of cell injury and death [49,50]. These reports suggest that kidney tubular cell injury and death promote interstitial fibrosis and inflammation. Since kidney tubular cell death is an early event that occurs before the onset of interstitial fibrosis and inflammation, it has been demonstrated that direct inhibition of caspase activation in a rat IRI model decreases tubular apoptotic cell death and prevents subsequent inflammation and fibrosis [51]. While NE prevents neuronal cell death from microglial inflammation and neurotoxicity [52,53], it induces caspase-3-dependent apoptosis in kidney tubule epithelial cells [27,28]. Our laboratory has reported that terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive kidney tubular apoptosis is diminished by renal denervation in mouse kidneys undergoing either UUO or IRI, whereas local infusion of NE into the denervated kidneys induces tubular apoptosis as a sequelae of kidney injury [27,28]. Cleaved forms of poly (ADP-ribose) polymerase 1 (PARP1) and caspase-3 are also increased by NE administration in cultured kidney proximal tubule epithelial cells (PTC), but co-treatment with a caspase-3 inhibitor significantly attenuates TUNEL-positive apoptosis induced by NE [27]. Our data are consistent with previous reports that NE induces apoptosis in neonatal cardiomyocytes and endothelial cells through caspase activation [54,55]. On the other hand, exogenous NE induces necrotic cell death in kidney proximal tubules in dogs [56,57]. Our recent in-vivo studies also supported a possible role for NE-induced necrotic cell death in mouse kidneys, characterized by increased PARP1 expression and tubular injury score in the denervated kidney undergoing either UUO or IRI [27,28]. However, the NE signaling pathway that induces apoptosis and necrosis in kidney tubular cells has not been delineated and requires further investigation.
Norepinephrine as a cell cycle arrester in fibrotic kidneys
Kidney tubular cell cycle arrest induced by severe AKI plays an important role in the development of fibrosis [58,59]. While cell cycle arrest is normally used as a protective mechanism to avoid cell division during stress and injury, sustained cell cycle arrest at the G2/M phase results in a senescence-associated secretory phenotype and leads to secretion of pro-proliferative and profibrotic factors such as connective tissue growth factor (CTGF) and TGF-β1, which can induce fibroblast proliferation and collagen deposition [60]. Our laboratory has reported that the tubular cell cycle arrest in the G2/M phase observed during interstitial fibrosis after IRI is prevented by renal denervation, as indicated by a decreased number of tubular cells positive for phosphorylated histone H3, and decreased ratio of cyclin B1 to cyclin D1, markers of the G2/M phase in the cell cycle [28]. However, NE infusion induces cell cycle arrest in the denervated kidney undergoing IRI [28]. Excluding the finding that G2/M-arrested kidney tubular cells activate the c-jun N-terminal kinase (JNK) signaling cascade that acts to upregulate profibrotic cytokine production [58], there is little information available about how cell cycle arrest links AKI to CKD. Despite no direct evidence showing that renal tubular cell cycle arrest mediates the AKI to CKD transition, profibrotic factors such as TGF-β1 derived from cell cycle arrested tubular cells may be indirectly involved in CKD progression, including fibrosis progression.
Norepinephrine can induce a profibrotic response in isolated renal proximal tubular cells
Our studies in cultured kidney PTC indicate that exposure to NE induces the production and secretion of TGF-β1 and CTGF [27]. TGF-β1, as a central mediator of fibrogenesis, induces CTGF upregulation through the binding of Smad3 to the CTGF promotor [61]. On the other hand, CTGF binds directly to TGF-β1, resulting in its increased activity through binding to two distinct TGF-β type I and II receptors [62]. Upon stimulation of TGF-β1 and/or CTGF alone, fibroblasts are activated and undergo a phenotypic transition into myofibroblasts [62]. We previously showed that culture medium from NE-exposed tubular cells, which contains released TGF-β1 and CTGF, triggers differentiation of kidney interstitial fibroblasts into α-SMA-positive myofibroblasts [27]. The myofibroblast is an activated form of fibroblast that is widely recognized as a major type of extracellular matrix-producing cell, and originates from bone marrow-derived cells or resident fibroblasts in fibrotic kidneys [63–65]. Although under debate, it is thought that kidney tubular cells also produce extracellular matrix, including fibronectin and collagen, and further undergo a phenotypic conversion into extracellular matrix-producing fibroblasts and myofibroblasts, the so-called epithelial-mesenchymal transition (EMT), during interstitial fibrogenesis after kidney injury [64,66,67]. Our laboratory has also shown that kidney proximal tubular cells exposed to NE can release fibronectin to the culture media independent of the TGF-β1 signaling pathway, but not EMT, as α-SMA expression was not detected in NE-treated PTC cells [27]. Therefore, NE-exposed kidney tubular cells contribute to interstitial fibrosis not only by stimulating extracellular matrix deposition derived from adjacent fibroblasts but also from themselves, suggesting that NE functions as a profibrotic inducer in kidney tubular cells.
Targeting the α2-adrenergic receptor in chronic kidney diseases
We previously demonstrated that inhibition of α2-ARs prevents interstitial fibrogenesis after IRI, as indicated by reduced TGF-β1 production, Smad3 phosphorylation, downregulation of α-SMA, and collagen deposition [28]. Other reports have indicated that inhibition of either α1-AR or β-AR protects kidneys against 5/6 nephrectomy-induced injury, and a combinational inhibition of α1-AR and β-AR is more effective in preventing glomerular, interstitial, and vascular injury than the inhibition of α1-AR or β-AR alone [68,69]. Our data implicating NE signaling through α2-AR in induction of fibrogenesis in IRI kidneys are intriguing, as presynaptic α2A-AR and α2C-AR subtypes in the vas deferens, isolated brain, and atrial tissue [70,71] and the α2A-AR subtype in the kidney play a predominant role in regulating synaptic NE release [72]. Loss of α2A-AR and α2C-AR subtypes increases susceptibility to development of heart failure after chronic pressure overload in mice [73,74].
Intriguingly, other investigators have reported that either activation of α2-AR using clonidine [75] and moxonidine [76] or inhibition of β-AR using propranolol [77] is protective against IRI. However, we found that the activation of α2-AR is detrimental and promotes the development and progression of fibrosis, inflammation, cell death, and cell cycle arrest after IRI [28]. Thus, the α2-AR activation in IRI-induced fibrotic kidneys may trigger alternate signaling events to sympathetic inhibition, such as activation of signaling pathways implicated in inflammation, cell death, and cell cycle arrest to instigate kidney fibrogenesis. This notion is supported by our finding that NE infusion in denervated kidneys enhances PMN-positive neutrophil and F4/80-positive macrophage infiltration, increases TUNEL-positive tubular apoptotic cell death, and induces cell cycle arrest in G2/M positive for phosphorylated histone H3 [28]. In UUO-subjected kidneys, inhibition of α2-AR using corresponding antagonists has no effect on NE level, but reduces cytokine/ chemokine expression [27], suggesting a possible mechanism by which NE may promote leukocyte recruitment and inflammation. This premise is supported by other reports demonstrating that α2-AR signaling activation using NE and UK-14304, an α2-AR agonist, augments the production of inflammatory cytokines (including TNF-α in macrophages) [78], accelerates TUNEL-positive apoptosis in mesenchymal cells [79], and triggers cell cycle arrest in oligodendrocyte progenitors [80].
Interaction between renal sympathetic nerves and RAS is highly associated with BP regulation and CKD progression [81]. Renal sympathetic nerve activation triggers renin release from the juxtaglomerular apparatus, which in turn results in an increase of angiotensin II (Ang II), the main effector of RAS and determinant of renal damage [81,82]. Conversely, Ang II can enhance NE level by acting on sympathetic nerve terminals, resulting in sympathoexcitation [19,83,84]. Hoch et al [85] showed that genetic inhibition of α2A-AR or pharmacological inhibition of α2-AR diminishes Ang II-mediated NE release in kidneys with 5/6 nephrectomy. In a recent report, Eriguchi et al [86] demonstrated that renal denervation halts CKD progression independent of BP in a rat model of Nω-nitro-L-arginine methyl ester (L-NAME; a nitric oxide synthase inhibitor), in which a hydralazine-mediated BP lowering effect had only a minor effect on preventing CKD progression, including kidney fibrosis, compared to renal denervation. The effect of renal denervation is associated with suppressed expression of intrarenal RAS components, indicating sympathetic regulation of intrarenal RAS. However, it remains to be defined whether renal sympathetic nerves or related signaling control intrarenal RAS in CKD progression through α2-AR.
In summary, intrarenal change following renal injury signals the central nervous system through renal afferents, and then the signal from the central nervous system contributes to sympathetic nerve activation and increases in renal NE level (Fig. 1). The renal sympathetic nerve-derived factor NE mediates the fibrogenic response, and α2-AR inhibition can prevent UUO- and IRI-induced renal interstitial fibrogenesis. These data are significant in that they suggest α2-AR as a primary signaling component that in turn regulates several of the key pathogenic molecules and processes implicated in renal inflammation, interstitial fibrogenesis and CKD (Fig. 1). Further, these findings are expected to have clinical translational potential, given that α2-AR inhibitors are already in clinical use for some diseases and in trials for other diseases, making them adaptable to prevent fibroproliferative diseases in the kidney and plausibly in other organs such as the liver, lung and heart.
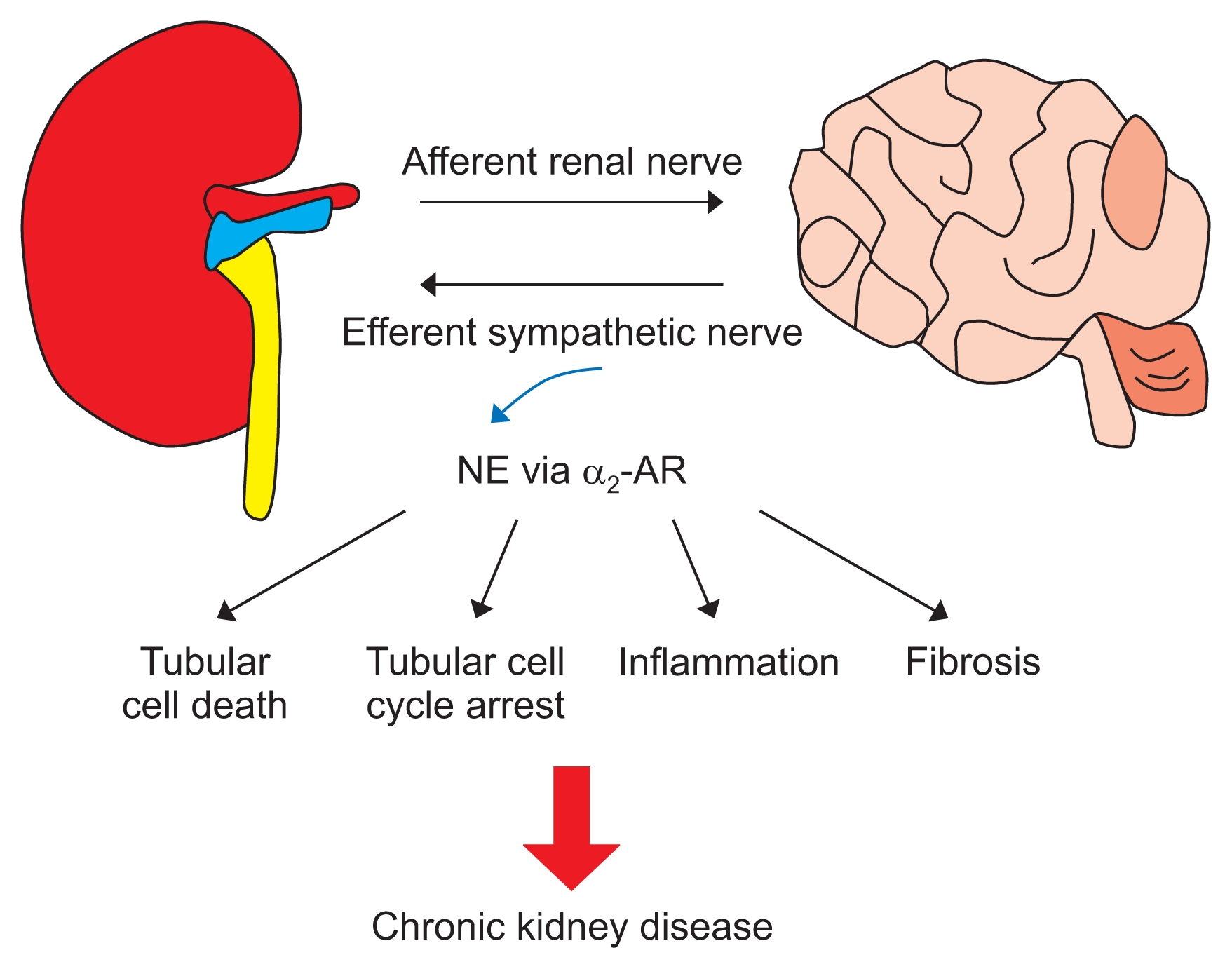
Renal sympathetic nerve-derived norepinephrine (NE) and alpha 2 adrenergic receptor (α2-AR) in chronic kidney disease development and progression
Intrarenal changes following renal injury, ischemia/reperfusion injury or unilateral ureteral obstruction are sensed by renal afferents, and integration of these signals in the brain contributes to sympathoexcitation and augments the sympathetic outflow and increase of renal norepinephrine level. The increased norepinephrine may trigger tubular cell death and cell cycle arrest, renal inflammation, and fibrosis progression through α2A- or α2C-AR, leading to chronic kidney disease.
Acknowledgments
This study was supported by NIH grants DK-083291 and DK-090332, American Heart Association (AHA) Grant in Aid 15GRNT25080031 (BJP) and AHA postdoctoral fellowship Grant 15POST25130003 (HSJ). J. Kim was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2016R1C1B2012080).
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.