


Introduction
Proteinuria is a common finding in many kidney diseases such as diabetic nephropathy and chronic glomerulonephritis. Elevated urine protein is strongly associated with glomerulosclerosis, interstitial fibrosis, and progression of chronic kidney disease [1], and independently predicts worse clinical outcomes [2]. Most agents that reduce proteinuria have beneficial effects on long-term renal function [3]. Despite widespread use of drugs that decrease proteinuria, progression to end-stage kidney disease remains all too common.
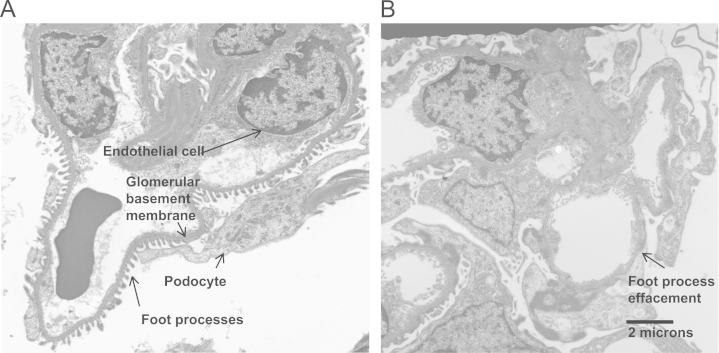
Traditionally, our understanding of the pathogenesis of proteinuria focused on glomerular hypertension. Effective treatment has been aimed at the reninŌĆōangiotensin system, which reduces intraglomerular pressure [4]. Over the past decade, interest has shifted from this mechanical view to a cellular and molecular perspective. The glomerular filtration barrier consists of endothelial cells, a basement membrane, and a specialized epithelial cellŌĆöthe podocyte (Fig. 1). Dysfunction in any component of the glomerular filtration barrier can lead to proteinuria. Increasingly, it has been recognized that there is a basal cross-talk between the endothelium and the podocytes, mainly involving angiogenic molecules such as vascular endothelial growth factor (VEGF) that maintain the health of the filtration barrier [5]. Perturbation of this signaling can lead to endothelial cell or podocyte injury. Podocyte injury manifests as retraction of the podocyte foot processes leading to effacement, loss of slit diaphragm proteins [6], dedifferentiation, detachment, and apoptosis [7]. Podocyte de-differentiation has been described as a process similar to epithelial to mesenchymal transition (EMT) and has been shown to occur in response to transforming growth factor-beta (TGF-╬▓) [8].
Causes and consequences of proteinuria
Many common kidney diseases such as diabetes and chronic glomerulonephritis first manifest with structural glomerular injury and proteinuria [9] followed by tubulointerstitial fibrosis [10]. Proteinuria is a prognostic indicator in a variety of kidney diseases. In a large cohort study, Hemmelgarn et al. [2] demonstrated that the presence of even mild proteinuria (trace to 1+ on urine dipstick or 30ŌĆō300┬Āmg/g albumin to creatinine ratio) increased the risk of mortality and progressive renal failure independently of baseline renal function and other prognostic markers. Proteinuria is a common condition with significant implications with respect to population health. In the National Health and Nutrition Examination Survey study, 8.2% of the general US population have mild proteinuria, and 1.1% have overt proteinuria [11].
Although proteinuria is strongly linked epidemiologically to progressive chronic kidney disease, there is ongoing debate as to whether proteinuria is causally linked to tubulointerstitial fibrosis. When renal tubular cells are grown in culture and exposed to high levels of protein, the cells express inflammatory and fibrogenic cytokines [12]. These in vitro findings are supported by evidence from animal models where protein overload alone induces chronic renal injury [13]. Other hypothesized mechanisms to explain progressive interstitial fibrosis after glomerular injury include loss of peritubular capillaries and tubular hypoxia [14], [15], filtered cytokines [13], misdirected filtrate from sclerosed glomeruli [14], and interstitial inflammation [15].
Although there is some disagreement [16], the prevalent paradigm is that proteinuria is primarily a result of dysfunction of the glomerular filtration apparatus with a special focus on the podocyte. The podocyte is a terminally differentiated epithelial cell with a highly specialized structure that consists of foot processes that wrap around the glomerular capillaries. A series of transmembrane proteins link the foot processes, and comprise the ŌĆśslit diaphragmŌĆÖ structure that is crucial to proper function of this barrier [6].
If podocytes are injured, the expression of the slit diaphragm proteins decreases. For example, in diabetic nephropathy, the expression of nephrin decreases [17] and nephrin appears in the urine [18]. Later in the course of glomerular disease, podocytes detach from the basement membrane and also are found in the urine [19], [20]. Podocyte apoptosis is also a common finding in advanced glomerular injury [21].
Communication between components of the filtration barrier
It is increasingly clear that components of the glomerular filtration barrier interact with each other directly or through paracrine mediators (Fig. 2). For example, podocytes secrete vascular growth factors that maintain glomerular endothelial health [22]. Elevated levels of podocyte derived VEGF leads to a collapsing glomerulopathy, whereas decreased VEGF leads to endotheliosis (endothelial swelling and fibrin deposition) [22]. An anti-VEGF antibody used commonly in bowel and renal cancer has been shown to increase the risk for proteinuria and hypertension [23], suggesting a direct clinical effect of altering the podocyte/endothelial cell interaction.
Glomerular endothelial cells show evidence of dysfunction at an early stage of glomerular injury [24] and secrete TGF-╬▓ in response to VEGF stimulation [25]. Paracrine interaction between endothelium and podocytes involves other angiogenic factors such as angiopoietin- (ANGPT) 1 [26] and ANGPT-2 [27]. These are secreted by podocytes and impact on endothelial cells.
Although not directly part of the glomerular filtration apparatus, mesangial cells play an important role in paracrine signaling. Mesangial cells respond to high glucose [28] and pathogenic immunoglobulin A (IgA) [29] by secreting TGF-╬▓. Khan et al. [30] have shown that mesangial cells can have a protective effect. TGF-╬▓ bound to the integrin ╬▒v╬▓8 is sequestered on mesangial cell membrane. When ╬▓8 integrin was knocked out, they observed higher levels of bioactive TGF-╬▓, which resulted in endothelial cell apoptosis and proteinuria [30]. Mesangial cells also produce other paracrine signals such as stromal-derived factor-1, which affects podocyte function [31].
Integrins play a role in the interaction between podocytes and the glomerular basement membrane (GBM). Integrin ╬▒3╬▓1 on podocytes allows adhesion with ╬▓2 laminin of the GBM. Interruption of this interaction leads to podocyte loss and glomerulosclerosis [32].
Role of angiopoietins in proteinuria
Angiopoietins are known to modify the effects of VEGF [33]. In quiescent vasculature, ANGPT1 is produced by perivascular cells and has a paracrine effect on endothelial cells. ANGPT1 binds to its receptor, tyrosine endothelial kinase (TEK), and signals through phosphatidylinositol-4,5 bisphosphate 3-kinase (PI3K)/Akt to maintain vascular integrity [34]. ANGPT2 similarly binds to the TEK receptor and is thought to be an antagonist for ANGPT1. In pathologic settings, VEGF and ANGPT2 are upregulated and lead to vascular destabilization, angiogenic sprouting, and new vessel formation [35]. In the quiescent glomerulus, ANGPT1 and VEGF are produced by podocytes and have protective and antiproteinuric effects on endothelial cells [26].
ANGPT2 has been associated with proteinuria and renal injury in several settings, including lupus nephritis [36] and diabetic nephropathy [4]. In previous work, Davis et al. [27] created a mouse model where ANGPT2 was overexpressed by a podocyte specific promoter. In this model, ANGPT2 overexpression led to proteinuria and decreased podocyte nephrin expression. The authors did not describe podocytes foot process effacement in response to ANGPT2, but they demonstrated endothelial cell dysfunction which was believed to underlie the proteinuric response.
In a recently described model of TGF-╬▓ overexpression in the glomerulus using an adenovirus [37], we found that ANGPT1 and ANGPT2 were both upregulated in the glomeruli from animals exposed to AdTGFB1, which is similar to observations by Campean et al. [38] in the Thy 1.1 model of glomerular injury. In unstimulated podocytes, we found no expression of TEK, but exposure to TGF-╬▓ increased TEK expression. We were able to block the effects of TGF-╬▓ on podocytes in culture with small interfering RNA directed against TEK, or with antibodies directed against TEK or ANGPT1. We hypothesized that TGF-╬▓ is required for podocyte dedifferentiation either as a direct cofactor or through the induction of TEK receptors on podocytes [37].
Transforming growth factor-beta
Much more is known about TGF-╬▓ than was known in late 1970┬Ās when De Larco and Todaro [39] considered it a ŌĆ£factorŌĆØ responsible for changing a cell's phenotype and called it ŌĆ£sarcoma growth factorŌĆØ. TGF-╬▓ has three isoforms in humans [40]. TGF-╬▓ is ubiquitously expressed and all cell types and all cells appear to respond to TGF-╬▓ [41]. In the glomerulus, TGF-╬▓ is produced by the podocytes and mesangial cells [42]. TGF-╬▓ is bound to a latency-associated peptide and sequestered in the extracellular matrix. Activation of TGF-╬▓ B is a complex and tightly regulated process [41], [43]. The mechanism of TGF-╬▓ activity at the cellular level is also complex, but the SMAD proteins are key signaling molecules for TGF-╬▓ [44].
TGF-╬▓ is involved in proteinuria and glomerulosclerosis
TGF-╬▓ is associated with fibrosis in many organ systems [45] including the kidney, where it can be induced by angiotensin [46], proteinuria [47], and hypoxia [48]. TGF-╬▓ induces elaboration of extracellular matrix components. Although controversial [49], TGF-╬▓ possibly induces EMT of renal tubular cells [50], [51]. Numerous studies have identified TGF-╬▓ as being upregulated during the course of progressive renal injury [52]. Similarly, inhibition of TGF-╬▓ has been shown to ameliorate renal injury [53].
TGF-╬▓ is not only involved in ECM accumulation, fibrosis, and progressive renal impairment, but also plays a role in changes to the glomerular filtration barrier and induction of proteinuria. There is substantial evidence to support this latter observation. Urinary TGF-╬▓ correlates with the degree of proteinuria [54]. Direct inhibition of TGF-╬▓ in models of renal disease reduces proteinuria [55], [56], [57].
Proteinuria was described in three animal models of TGF-╬▓ overexpression [58], [59], [60]. High levels of systemic TGF-╬▓ using a transgenic mouse model were shown to induce proteinuria, but the mechanism was not described [58]. Krag et al. [59] described a novel transgenic mouse with TGF-╬▓ driven from the Ren-1c promoter of the juxtaglomerular apparatus that developed albuminuria [59]. Wang et al. [60] used a hydrodynamic gene transfer model to overexpress TGF-╬▓ in the glomerulus and identified the role of Wnt/╬▓-catenin in TGF-╬▓-induced proteinuria.
TGF-╬▓ and podocyte dedifferentiation
The presence of the slit diaphragm as an important component of the glomerular filtration apparatus has been known for more than 30 years. Over the past 15 years, key molecular components of the slit diaphragm such as nephrin have been identified [61], the loss of which is associated with proteinuria [6]. Initially, this loss of epithelial markers was termed ŌĆ£de-differentiationŌĆØ [62], but work by Sam et al. [63] later supported by Li et al. [8] identified that podocytes may undergo EMT in response to TGF-╬▓.
Podocyte dedifferentiation is an early event in glomerular injury, whereas subsequent podocyte apoptosis and sloughing has been observed in advanced renal disease [7]. Notably, apoptosis appears to be a later finding that occurs after the onset of proteinuria [21]. Podocyte cell death is a complex process involving not only apoptosis, but also anoikis, autophagy, and, in some cases, necrosis [64].
Several pathways and growth factors are involved in this EMT process. As mentioned, TGF-╬▓ has been shown to induce podocyte EMT in vitro
[8], [63], [65]. SNAIL is one of a family of zinc finger regulatory proteins shown to be an important mediator of EMT [66] and has been shown to suppress nephrin gene transcription in a model of puramycin-induced nephrosis [67]. Other pathways shown to be involved in podocyte EMT include mammalian target of rapamycin (mTOR) [68], [69], glycogen synthase kinase 3 ╬▓ (GSK3B) [70], ╬▓-catenin [71], integrin-linked kinase (ILK) [72], and Notch [73]. PI3K has also been shown to be a key regulator of podocyte EMT, and PI3K inhibition was shown to block podocyte injury in an in vitro IgA model [74]. Recently, Finer et al. [75] have shown that podocyte injury induced by Adriamycin is mostly mediated by PI3K, and TGF-╬▓ is not involved in this process, but is involved in a delayed, fibrogenic response.
Notch is proving to be a key regulator of podocyte injury and apoptosis. Notch is a transmembrane receptor protein. On ligand binding, Notch undergoes a series of modifications including intracellular cleavage by gamma secretase [76]. The intracellular Notch fragment then acts as a transcriptional regulator. Notch interacts with the ╬▓-catenin and Wnt pathways during development of the kidney, but is quiescent in the adult kidney. During injury, TGF-╬▓ increases expression of the Notch ligand Jagged 1 and thus induces Notch signaling. In a seminal paper, Niranjan et al. [77] showed that overexpression of the intracellular Notch fragment in podocytes leads to severe proteinuria, glomerulosclerosis, and podocyte apoptosis. Using a gamma secretase inhibitor to block Notch signaling, renal injury could be reduced in a rodent model [77].
TGF-╬▓ mainly employs SMAD proteins to translate its signal; however, other signaling pathways have been shown to be activated by TGF-╬▓. We have shown that in peritoneal mesothelial cells, EMT still occurs in mice lacking SMAD3, suggesting that non-SMAD pathways are critical for EMT [78]. This is supported by the observation of Wang et al. [79], who observed ongoing proteinuria and foot process effacement in SMAD3ŌĆō/ŌĆō mice rendered diabetic. The SMAD3ŌĆō/ŌĆō mice were protected from glomerulosclerosis and renal dysfunction.
EMT has been observed in urinary podocytes in patients with diabetes [80]. Although understanding podocyte EMT will provide valuable insights into the mechanisms of early glomerular injury progressive kidney disease, there are also therapeutic implications. For example, it has been demonstrated that TGF-╬▓ antagonists, specifically bone morphogenetic protein 7, can reverse EMT changes in renal tubular epithelium [50] and podocytes [81].
TGF-╬▓ and glomerular endothelial cells
Research into the biology of glomerular endothelial cells has perhaps been overshadowed by a strong interest in the podocyte. Early research suggested that the size of endothelial cells fenestrations is much larger than most plasma proteins (60ŌĆō100┬Ānm) and thus the endothelium should not form a significant filtration barrier. More recent work identified the importance of the glycocalyx, a dense negatively charged protein complex, as an important component of the glomerular filtration barrier [82]. Any injury that perturbs the integrity of this glycocalyx may lead to proteinuria [83].
Podocyte-derived VEGF has direct action on glomerular endothelial cells including proliferation and production of TGF-╬▓ [25]. VEGF also appears to regulate endothelial fenestrations [22]. Endothelial fenestrations were decreased in a biopsy study of Pima Indians with diabetes [19]. This decreased endothelial fenestration correlated with decreased glomerular filtration and increased albuminuria, whereas podocyte loss correlated with albumin excretion alone. There has been a direct association observed between podocyte injury and endothelial injury possibly mediated by loss of podocyte VEGF excretion [84].
Glomerular endothelial cells also respond to angiopoietins. Satchell et al. [26] have demonstrated that glomerular endothelial cells express the angiopoietin receptor TEK. In this study, ANGPT1 decreased protein passage through endothelial cell monolayers. As mentioned previously, overexpression of ANGPT2 in podocytes led to endothelial apoptosis and proteinuria [27]. El-Banawy et al. [85] measured ANGPT2 in patients with lupus and found that serum concentration correlated with proteinuria and disease severity.
A recently identified mechanism by which TGF-╬▓ induces endothelial cell dysfunction is endothelial to mesenchymal transition [86]. This is a similar biological process to EMT, and involves many of the same initiating factors and signaling pathways [86]. These transitioned endothelial cells are demonstrated to be the source of interstitial fibrosis in three well-known mouse models [87]. Although there is reasonable evidence that endothelial mesenchymal transition plays a role in interstitial fibrosis, few studies have evaluated glomerular endothelial cells. In one such study of streptozotocin-induced diabetic nephropathy, Li et al. [88] used an endothelial lineage traceable transgenic mouse to show that endothelial mesenchymal transition does occur in glomerular endothelial cells. However, this observation predated the onset of proteinuria in this model, so the authors thought that endothelial transition may play a role in glomerulosclerosis, but not proteinuria. This same study identified TGF-╬▓ as an in vitro mediator of endothelial mesenchymal transition [88].
TGF-╬▓ and the GBM
The GBM is a unique structure composed of laminin, type IV collagen, nidogen, and heparan sulfate proteoglycan. Both podocytes and endothelial cells have a role in development and maintenance of this structure [89]. Early changes in diabetic nephropathy include thickening of the GBM [90]. Krag et al. [91] have shown that mild hyperglycemia alone does not affect the GBM, but combined hyperglycemia and overexpression of TGF-╬▓ leads to pronounced GBM thickening. Wang et al. [79] found that the diabetes-induced GBM thickening was attenuated in mice deficient in the TGF-╬▓ signaling protein SMAD3.
The GBM is clearly an integral component of the glomerular filtration barrier; several genetic diseases that manifest as proteinuria have defects in proteins expressed in the GBM [89]. The structure and composition of the GBM can be altered by TGF-╬▓, and this can potentially lead to further changes in the glomerular filtration barrier and proteinuria.
Therapeutic significance
The evidence implicating TGF-╬▓ B in the induction of proteinuria, glomerulosclerosis, and renal fibrosis is compelling. This makes TGF-╬▓ an important target in the prevention of progressive renal disease. Direct inhibition of TGF-╬▓ is a potential therapeutic strategy and a recent Phase 1 clinical trial of an anti-TGF-╬▓ antibody showed that a single infusion of this agent was well tolerated [92].
Many conditions that lead to end stage kidney disease are chronic processes that would necessitate long term inhibition of TGF-╬▓. TGF-╬▓ is pleiotropic and has roles in wound healing, inflammation [93], and suppression of early cancers [94]. Long-term inhibition of TGF-╬▓ likely carries significant risks. Downstream pathways activated by TGF-╬▓ have become important therapeutic targets. For example, connective tissue growth factor is induced by TGF-╬▓ and is upregulated in patients with diabetic nephropathy [95]. In a Phase I clinical trial, the anti-connective tissue growth factor antibody FG-3019 was well tolerated and demonstrated a significant reduction in albuminuria in patients with diabetic nephropathy [96].
Another potential downstream target of TGF-╬▓ is the mammalian target of rapamycin (mTOR). We have demonstrated that mTOR is important in TGF-╬▓ -induced EMT, especially in the absence of a SMAD3 signal [78]. Although associated with increased risk of proteinuria in renal transplantation, recent studies have suggested that low-dose mTOR inhibition may have anti-proteinuric effects, at least in certain animal models [97].
TGF-╬▓ has been shown to activate the Wnt signaling pathway in podocytes [60]. Blockade of Wnt signaling in vivo blocked downstream signals involved in EMT such as SNAIL, and reduced TGF-╬▓ induced urinary protein excretion. Canonical Wnt signaling, through facilitation of ╬▓-catenin action, have been implicated in fibrosis of various organ systems [98] and treatment with a compound that specifically inhibits ╬▓-catenin (ICG-001) has been effective in animal models of lung and kidney fibrosis [99], [100].
Increasingly, both genetic and epigenetic processes have been identified in fibrogenesis [101]. Micro RNAs (miRNAs) are small, noncoding RNA fragments that modulate the expression of other coding mRNAs. TGF-╬▓ has been shown to increase the expression of miRNA 192 which then alters downstream signaling events. Blocking miRNA has recently been shown to have a beneficial effect in a mouse model of diabetic nephropathy [102]. Specifically, blocking miR-192 reduced albuminuria and renal fibrosis.
Summary
Proteinuria is a harbinger of progressive renal dysfunction in a wide range of common kidney diseases. Even low levels of proteinuria are associated with increased morbidity, mortality, and progression to end-stage renal disease [2]. Therapies, such as angiotensin pathway inhibitors, have been shown to reduce proteinuria and slow down the progressive decline in renal function. Despite this, further therapies are required. There is increasing evidence that TGF-╬▓ plays an important role in induction of proteinuria, along with subsequent glomerulosclerosis and renal interstitial fibrosis. Directly targeting TGF-╬▓ is an exciting approach to chronic renal disease. Further understanding of the TGF-╬▓ pathways involved will lead to a range of novel therapies that will reduce proteinuria and prevent progressive decline in renal function.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









