T helper 17 cells in the pathophysiology of acute and chronic kidney disease
Article information

Abstract
Both acute and chronic kidney disease have a strong underlying inflammatory component. This review focuses primarily on T helper 17 (Th17) cells as mediators of inflammation and their potential to modulate acute and chronic kidney disease. We provide updated information on factors and signaling pathways that promote Th17 cell differentiation with specific reference to kidney disease. We highlight numerous clinical studies that have investigated Th17 cells in the setting of human kidney disease and provide updated summaries from various experimental animal models of kidney disease indicating an important role for Th17 cells in renal fibrosis and hypertension. We focus on the pleiotropic effects of Th17 cells in different renal cell types as potentially relevant to the pathogenesis of kidney disease. Finally, we highlight studies that present contrasting roles for Th17 cells in kidney disease progression.
Background
As of 2017, the global burden of chronic kidney disease (CKD) was estimated to be approximately 9.1%, indicating that CKD is a significant healthcare concern worldwide. Global age-adjusted mortality rates for patients with CKD have declined in the last 30 years, and quality of life metrics decrease as glomerular filtration rate declines [1]. In the period between1990 and 2017, there was an approximate 41% increase in the number of patients with end-stage renal disease (ESRD). Despite this, CKD patients are more likely to die due to complications such as hypertension, cardiovascular disease, anemia, and bone and mineral disorders, and these patients also have an increased cancer incidence [1].
Acute kidney injury (AKI), generally defined as a rapid loss of kidney function, results in renal damage caused by factors such as nephrotoxins, radiocontrast agents, hypoperfusion, surgery, sepsis, and antibiotics [2]. AKI occurs in up to 7% of hospital admissions per year in the United States [2–4], while a recent study based on KDIGO (Kidney Disease: Improving Global Outcomes) guidelines suggested that up to 1 in 5 adult hospitalized patients worldwide have some form of AKI [5]. AKI is associated with significant morbidity and mortality [4,6,7], and surviving patients are at increased risk of developing CKD and ESRD [8].
Both acute and CKD have a strong underlying inflammatory component. The purpose of this review is to focus primarily on T helper 17 (Th17) cells as mediators of inflammation and present evidence that these cells modulate both acute and CKD and that the primary cytokine produced by these cells, interleukin (IL) 17A, is a potential therapeutic target in kidney disease.
T helper 17 definition and roles
CD4+ T cells have pleiotropic potential and are thought to assist in the humoral immune response indirectly by affecting innate immune cells such as neutrophils and macrophages. T-cell activation occurs when T-cell receptors (TCRs) recognize specific peptides on MHCII+ antigen presenting cells (APCs). Coupled with co-stimulatory signals derived from APCs, as well as cytokines and other signals from innate immune cells, these combined inputs drive T-cell differentiation and expansion. In 1986, Mosmann et al. [9] defined two distinct sets of T-helper cells based on their lymphokine profiles; Th1 and Th2 cells. The transcription factors T-bet and STAT-1 are essential for the differentiation of Th1 cells, characterized by the secretion of interferon (IFN)-γ, IL-2, IL-12, and tumor necrosis factor (TNF)-α. The primary function of Th1 cells is to assist in the macrophage response against intracellular pathogens such as viruses or bacteria [10]. In contrast, STAT-6 and GATA3 drive differentiation of Th2 cells, characterized by the secretion of IL-4 as well as IL-10, IL-13, and IL-25 [11]. A primary function of Th2 cells in immune responses is to fight extracellular parasites by regulating mucous production in the gastrointestinal tract and stimulating antibody production to facilitate destruction of invading parasites by eosinophils or mast cells [10].
In 2005, two groups described a population of T-helper cells referred to as Th17 cells based on the expression of the signature cytokine IL-17A. These cells also secrete other factors such as IL-17F, IL-21, IL-22, IL-23, and TNF-α [12–14]. Th17 cells play a critical role in host protection against certain types of pathogens such as extracellular bacteria, which are not traditional targets of Th1 or Th2 cells [10]. Under the influence of IL-17A, Th17 cells are strongly proinflammatory, resulting in an influx of neutrophils to fight infections [14].
Like Th1 and Th2 cells, Th17 cells differentiate from naïve Th0 cells following engagement of TCRs and the influence of local cytokines (reviewed in [15,16]). This response is dependent on the expression of RAR-related orphan receptor gamma T (RORγt), the key transcriptional regulator of Th17 cell differentiation. This process is dependent on the proinflammatory cytokine IL-6 and the activation of STAT3. Other proinflammatory cytokines, such as IL-21, can also activate STAT3 to induce expression of RORγt (Fig. 1). In addition, transcription factors such as basic leucine zipper ATF-like transcription factor or interferon regulatory factor 4 can modulate the chromatin accessibility of Th17 specific genes and act cooperatively with RORγt to modulate Th17 differentiation [17]. Th17 differentiation is also thought to be dependent on the activity of transforming growth factor (TGF)-β, since TGFβ null mice, or mice that express a dominant negative form of TβRII on CD4+ cells, lack the ability to produce mature Th17 cells, while mice overexpressing TGF-β display enhanced Th17 activation [16].
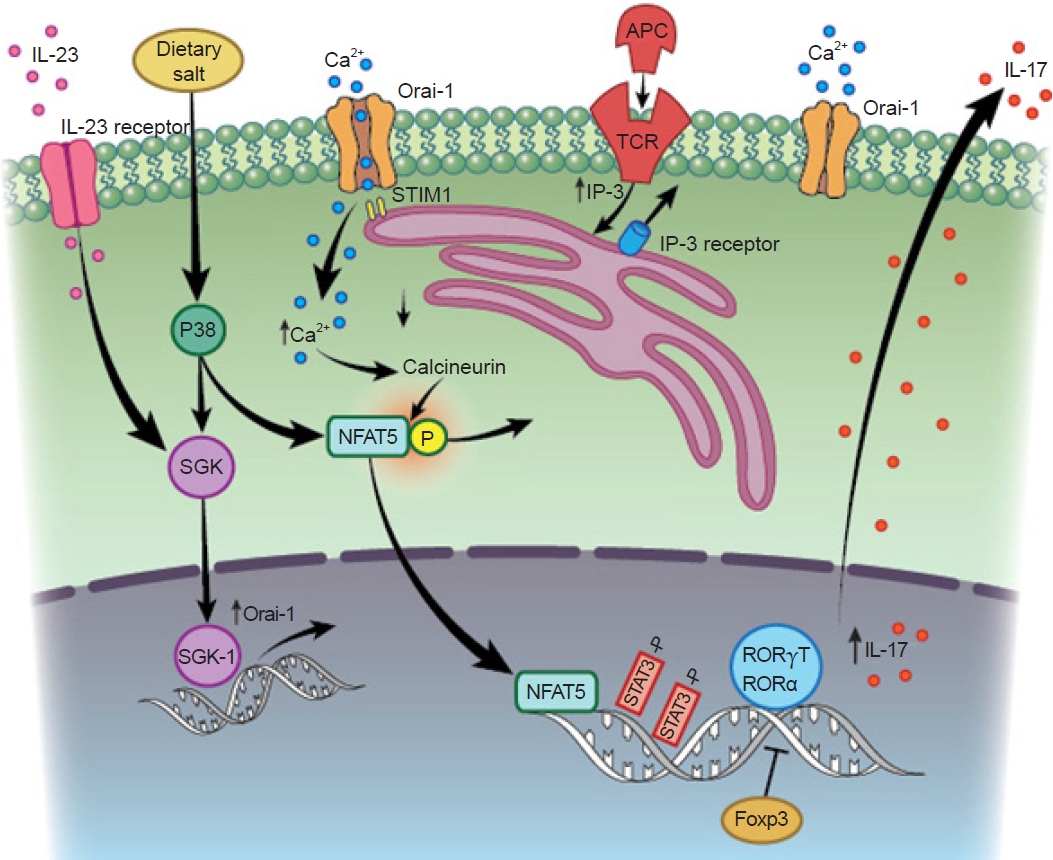
Activation and differentiation of T helper 17 (Th17) cells in the setting of renal injury and potential influence of salt.
Th17 cell differentiation and interleukin (IL) 17 activation occur secondary to T-cell receptor (TCR) activation. Activation of STAT3 is required for induction of the transcription factor RAR-related orphan receptor gamma T (RORγT), which mediates transcription of the signature cytokine, IL-17. The process appears to be dependent on activation of the store-operated calcium release activated calcium (CRAC) channel Oria-1, which is initially activated by stromal interaction molecule 1 (STIM1) following endoplasmic reticulum depletion of Ca2+. The sustained elevation of Ca2+ mediated by Oria1 activates nuclear factor of activated T cells (NFAT) dephosphorylation and further drives IL-17 expression. Oria1 is also activated by a high-salt environment, possibly resulting from serine/threonine-protein kinase (SGK) signaling, which may further amplify Th17 activity in the setting of elevated dietary sodium intake.
APC, antigen presenting cells; IP-3, inositol 1,4,5-trisphosphate.
Calcium signaling is induced following TCR stimulation, which is critical in driving the differentiation response. Several studies have indicated an important role for the store-operated calcium release activated calcium channel (CRAC) Oria1 in Th17 differentiation. Activation of this channel is dependent on depletion of endoplasmic reticulum stores of Ca2+ secondary to TCR activation, which is sensed by the endoplasmic reticulum transmembrane protein Stim1 and its subsequent interaction with plasma membrane Orai1 to mediate enhanced Ca2+ influx (Fig. 1) [18,19]. Mutations in either Orai1 or Stim1 result in a severe combined immunodeficiency phenotype [20]. Kim et al. [21] identified putative Orai1 inhibitors that showed greater selectivity in abrogating Th17 differentiation vs. Th1 or Th2 differentiation by screening a chemical library. Orai1 inhibition also reduced the nuclear accumulation of nuclear factor of activated T cells and RORγT. We suggested a requirement for Orai1 in Th17 differentiation in our recent study of a rat model of kidney injury; IL-17 expression by CD4+ cells was exclusive to those cells that expressed Orai1 and was not detected in CD4+ cells that lacked expression of this channel [22].
The IL-17 receptor family comprises five members; IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE. The primary ligands secreted by Th17 cells, IL-17A and IL-17F, form homodimers or IL-17A/F heterodimers and engage primarily with IL-17RA, but likely require complex formation with IL-17RC for efficient binding or signal transduction (for a detailed review see [23]). A complex pattern of differing affinities of IL-17A, IL-17F, and IL-17A/F for different receptors, as well as different ratios of IL-17RA and IL-17RC present in different cell types have been proposed to account for tissue-specific responses [24]. Importantly, Kuestner et al. [25] demonstrated that a soluble IL-17RC-Fc fusion protein was able to bind to both IL-17A and IL-17F, an approach that was shown to inhibit the activity of both of these ligands in vivo.
Role of interleukin 17 in kidney pathophysiology
IL-17 receptors are expressed in many different cell types but are prominent in cells of hematopoietic origin [24]. Because the kidney is vulnerable to infection, Th17 cells are thought to serve a critical role in antimicrobial host defense [26,27]. IL-17 is among a number of factors that are elevated in patients with acute urinary tract infections (UTIs) [28]. Increased expression of IL-17A was observed in the bladders of mice infected with uropathogenic Escherichia coli. Furthermore, IL-17A-/- mice have reduced macrophage and neutrophil influx, resulting in impaired clearance of bacterial load [29]. Interestingly, Olson et al. [30] demonstrated increased UTI severity in male mice vs. female mice and suggested that sex differences in UTI outcomes were due to a greater IL-17 response in females than males [31]. In a model of systemic Candida albicans infection, infected mice were shown to have increased IL-17 expression while null IL-17RA-/- mice showed increased renal fungal colonization, reduced neutrophil infiltration, and decreased survival [32]. An interesting study by Ramani et al. [33] demonstrated that tubular specific deletion of IL-17RA was essential for activation of host defenses in response to C. albicans infection, suggesting that IL-17 expressed by renal parenchymal cells plays a role in fighting infections.
Autoimmune and inflammatory diseases
While the Th17 system plays an important role in host-defense, unrestrained or inappropriate IL-17 signaling may exacerbate tissue damage due to its strong proinflammatory properties. Accumulating evidence suggests that Th17 cells or increased IL-17 expression is a common feature of many kidney diseases (Table 1 [22,28,34–59]). For example in the setting of autoimmune disease, although initially considered the result of Th1-mediated effects, hyper-activation of Th17 cells (or potentially other IL-17-secreting cells) has been suggested to exacerbate conditions such as psoriasis, inflammatory bowel disease, and autoimmune encephalitis [60].

Interleukin (IL) 17 expression in kidney diseases and hypertension
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by the production of auto-antibodies. Th17 cells have been proposed to mediate the interaction between immune complexes and alterations in kidney structure and function [61]. Lupus patients have been shown to have more circulating CD4+/IL-17+ cells than control patients [34], and circulating IL-17 levels were shown to be higher in patients with active rejection than those in a non-rejection state and were undetectable in healthy control patients [35]. Another study reported that baseline levels of IL-17 and IL-23 in SLE patients predicted a poor response to treatment [36].
Similarly, Krebs et al. [38] demonstrated higher levels of kidney RORγt+ T cells in patients with antineutrophil cytoplasmic antibody (ANCA)-associated glomerular nephritis. Patients with acute ANCA vasculitis had higher circulating IL-17 and IL-23 than healthy controls, and these levels remained persistently elevated above those in control patients even after inflammation resolved [40]. IL-17 immunostaining was identified in renal biopsies from patients with ANCA nephritis, although positively-stained cells were initially identified as polymorphonuclear granulocytes with lower frequencies of IL-17+ T cells [39]. Both serum and urinary levels of IL-17 have been shown to be elevated in patients with immunoglobulin A (IgA)-nephropathy (IgAN) [41], another immune complex-mediated disease triggered by mesangial deposition of IgA leading to CKD.
Data from multiple mouse models support the hypothesis that Th17 cells contribute to inflammatory disease progression. These mouse models include crescentic glomerular nephritis following myeloperoxidase injection [62,63]; exposure to the Goodpasture antigen, α3IV-NC1 [64]; lupus nephritis induced by pristane injection [65]; and injection of anti-glomerular basement membrane antibody [38]. Th17 cell accumulation in the inflamed kidneys was reported for all these models. Furthermore, reduced prevalence of kidney disease was observed in studies that used IL-17A-/- mice or rorγt-/- mice [66] relative to wild-type mice. The protection was associated with reduced infiltration of other cells such as neutrophils and macrophages and the expression of cytokines such as C-C motif chemokine ligand (CCL) 5.
Paust et al. [67] examined the dynamics of both Th1 and Th17 cells following injection of sheep serum into mice to induce glomerular inflammation. Th17 cells peaked in the earlier phase of the disease (i.e., 10 days following injection), while Th1 cells peaked during the later phase of the disease (>20 days). Null mutations or immunoneutralization of either IL-17 or IFNγ reduced the degree of renal dysfunction and tissue injury, while adoptive transfer of wild-type splenocytes, but not IL-17-/- or IFN-/- derived splenocytes, sutained the degree of renal damage in T-cell-deficient Rag1-/- mice. Interestingly, adoptive transfer of Th17 cells primed by glomerular antigens into Rag1-/- mice was sufficient to drive the formation of glomerular crescents [68].
Infiltration of Th17 cells into the kidney is due in part to local expression of cytokines. Using the MRL/lpr model of lupus nephritis, Steinmetz et al. [69] reported an influx of both Th1 and Th17 cells that appeared to be dependent on the activity of CXCR3 on CD4 cells, as mutation of this receptor reduced infiltration of both Th1 and Th17 cells. CXCR3 interacts with at least three ligands, Mig/CXCL9, IP-10/CXCL10, and ITAC/CXCL11, suggesting that these may play a role in Th17 infiltration. Lu et al. [70] demonstrated that incubation of human mesangial cells with serum from IgAN patients stimulated the production of CCL20. Through interaction with its cognate receptor chemokine receptor 6 (CCR6), CCL20 was shown to be chemo-attractant to IL-17+CD3+ T cells in transwell experiments. These IL-17+ T cells showed enhanced expression of CCR6, and immunofluorescence staining identified a population of CD3+IL-17+CCR6+ cells in kidneys of IgAN patients but not in control kidney samples [70]. In the study of Paust et al. [67], infiltration of Th17 cells was attributed to an early increase in the expression of CCL20. Using a method dependent on a photo-convertible fluorescent protein in Kaede mice, Th17 cells were shown to traffic from the gut to the kidney in response to ANCA exposure. The migration of Th17 cells was dependent on the activity of the CCL20/CCR6 axis, and the severity of the disease and infiltration of Th17 cells could be up- or down-modulated by treatments that affected resident gut T cells [38].
Complications in renal transplantation
The Th17 system has also been suggested to contribute to rejection of solid organ transplants. Acute allograft rejection is initiated by alloreactive T cells primed in secondary lymphoid organs and recruited to the graft. There is increasing evidence that IL-17 expression in local tissue is associated with allograft rejection [71,72]. In the setting of kidney transplantation, IL-17 was identified by immunohistochemistry in biopsies of patients with acute rejection compared with control patients, while levels of Foxp3, a marker of T-regulatory (Treg) cells that inhibits Th17 cells via the activity of IL-10, were reduced [46,47]. Chronic IL-17 expression in tubular epithelial cells was also shown to be associated with acute antibody-mediated rejection [45]. Some studies have suggested that outcomes in acute rejection are dependent on the degree of Th17 activation. For example, Millán et al. [48] showed that circulating IL-17 levels and the percentage of IFN-γ+CD4+CD69+ and IFN-γ+CD8+CD69+ cells could identify patients at high risk of acute rejection following either liver or kidney transplantation. Matignon et al. [49] demonstrated that elevated IL-17 messenger RNA (mRNA) in renal biopsies of elderly transplant recipients was associated with non-successful reversal of acute rejection. Recently, Nova-Lamperti et al. [73] demonstrated that tolerant recipients had fewer Th17 cells than patients with chronic rejection, while isolated peripheral blood mononuclear cells (PBMCs) from tolerant transplant recipients manifested reduced TCR signaling and Th17 differentiation compared to PBMCs from patients developing chronic rejection.
Evidence from animal models further supports a role for IL-17 in transplant rejection [46]. In models of cardiac or liver allograft rejection, increased expression of IL-17 by both CD4 and CD8 cells was observed, and an imbalance in Th17/Treg cells was shown to be associated with allograft rejection [74–76]. In a liver model of allograft transplantation, immunoneutralization of IL-17 improved expression of Treg cells and liver survival [75], while IL-17-/- mice exhibited increased Treg expression and survival following cardiac allograft transplant surgery [76].
Patients with chronic allograft nephropathy showed an increase in prevalence of Th17 cells following renal transplantation [50]. Mortazavi et al. [77] demonstrated that immunosuppressive therapy was associated with an improved Th17/Treg balance. Abadja et al. [78] demonstrated that mycophenolate has a stronger Th17 inhibitory effect than tacrolimus in in vitro activated human CD4 cells. In addition, these investigators showed that patients treated with both mycophenolate and low doses of tacrolimus had lower circulating levels of IL-17 than patients treated with tacrolimus alone. Low vitamin D levels have been shown to inversely correlate with Th17 cells in transplant recipients, and calcitriol supplementation was shown to suppress Th17 cells relative to treatment with tacrolimus alone [79]. These data suggest a role for Th17 cell responses in graft outcomes.
Responses in acute injury
As described above, T-lymphocytes as well as other immune cells are known to play roles in autoimmune and immune-mediated diseases as well as complications from renal transplant. However, other kidney disorders, such as acute injury, hypertensive and diabetic nephropathy, and the development of interstitial fibrosis, have a complicated pathophysiology that entails cellular responses to injury secondary to hypoxia, toxins, or elevated pressures, alterations in hemodynamics, activation of fibrogenic factors, and interstitial remodeling. Studies over the past few years have shown that the immune system plays a previously unappreciated role as a modulator of primary pathophysiological processes in these different types of kidney disease.
For example, AKI is a significant clinical disorder resulting from renal ischemia or nephrotoxic injury. While damage to the renal microvasculature or tubular epithelium is the primary component in the pathogenesis of AKI [2], the potential role of inflammation as a modulator of renal injury has received significant attention over the last 15 years [80]. Yokota et al. [81] published several seminal studies describing the importance of lymphocytes as mediators of renal injury. Studies from this group using STAT4-/- mice suggested a potential role for Th1 in the pathogenesis of AKI, while studies using STAT6-/- mice suggested a potential role for Th2-derived IL-4 in protection from renal injury [82].
Recently, the potential contribution of Th17 cells to AKI has been examined. Our group demonstrated an increase in circulating IL-17+ PBMC and Th17 (CD4+IL-17+) cells in patients of intensive care unit after diagnosis of AKI vs. critically ill patients without AKI [22], while Maravitsa et al. [51] reported that IL-17 levels were increased in septic patients that developed AKI. Using a rat model of renal ischemia/reperfusion (I/R) injury, our research group demonstrated that Th17 cells were the predominant T-helper subtype present in the kidney following acute injury, peaking between 1 to 3 days post I/R but remaining persistently elevated for several weeks following recovery from I/R. Worsening AKI in rats that were placed on a vitamin D-deficient diet was associated with an increased number of Th17 cells and a decreased number of Treg cells compared to rats on a standard diet [83]. Treatment with sIL-17RC or the store-operated Ca2+ entry channel (SOCC) inhibitor YM58483 blocked Th17 induction and significantly attenuated the development of renal injury [22,84]. Similar increases in Th17 cells in response to I/R injury have been reported in mice, while T-cell specific STAT3 deletion attenuated Th17 activation and resulted in protection against I/R [85]. In addition, rats with a mutation of the gene encoding RORγt showed impaired Th17 responses but not Th1 or Th2 responses, and were protected from renal injury by I/R [86]. In other models of AKI, such as the cecal-ligation puncture model of sepsis, IL-17-/- mice showed reduced renal cytokine expression, neutrophil infiltration, and tubular injury relative to wild-type mice [87]. Chan et al. [88] demonstrated reduced injury in response to cisplatin treatment in IL-17-/- mice and Rorγt-/- mice, while our group demonstrated that Th17 blockade protected against AKI in glycerol-induced rhabdomyolysis [22]. The influx of Th17 cells in the setting of AKI may be due in part to the activation of CCL20 [88]. In addition, a recent study suggests that damaged renal tubules produce IL-17C, which may recruit Th17 cells to promote inflammation via binding to the receptor IL-17RE [89]. The precise mechanisms by which Th17 cells contribute to renal injury are not clear, but are likely related to an overall inflammatory response leading to recruitment of effectors cells or contributions to vascular congestion and hypoxia.
Effective recovery from AKI is a result of a tissue repair response. However, incomplete or ineffective repair may predispose to the secondary development of CKD. The AKI-to-CKD transition may result from persistent alterations in vascular, tubular, and/or interstitial compartments [90]. Our group recently proposed a central role for persistent Th17 activity in this process. An AKI-to-CKD transition model was established by exposing rats to a high-salt diet after 5 weeks of recovery from I/R injury. This procedure, which hastened the development of inflammation, fibrosis, and hypertension, also potently stimulated the re-expression of Th17 cells [91]. CKD progression and hypertension in response to high-salt treatment was attenuated by mycophenolate, which also blocks induction of Th17 cells [84,92]. Th17 activation and CKD progression were also blocked by treatment with losartan or the SOCC inhibitor YM58483 [22,84]. Interestingly, the AKI-to-CKD transition in athymic nude rats, which lack conventional Th17-cells but display a compensatory increase in IL-17 production by natural killer cells (NK cells; CD3–/CD161+), was also blocked by sIL-17RC [84].
Chronic kidney disease, fibrosis, and hypertension
Biopsies of patients with renal fibrosis showed elevated renal IL-17 expression [52], while other studies have identified polymorphisms within the IL-17E and IL-17RA genes associated with ESRD [93]. CKD models associated with interstitial fibrosis, such as the unilateral ureteral obstruction (UUO) model [52,94-97] or adriamycin-induced nephropathy, are characterized by elevations in renal Th17 cells [98]. In all of these studies, strategies to neutralize of IL-17 activity, either with antibodies or by using IL-17-/- mice, mitigated the degree of renal fibrosis in these models. The fibrotic effect of IL-17 may be modulated by other cytokines. For example, the activity of the IL-17/IL-23 axis in inducing renal fibrosis was diminished in animals that lacked IL-36 receptors [94], while IL-27-/- mice showed increased Th17-induced fibrosis in response to UUO [99].
Diabetic nephropathy is known to have a strong inflammatory component and the role of Th17 cells in this setting has received significant attention in recent years [100]. Patients with type II diabetic nephropathy show a skew toward circulating Th1 and Th17 cells and have increased serum levels of IL-6 and IL-17, correlated with renal albumin excretion [54]. A recent study of both T1D and T2D patients demonstrated that IL-17A and IL-17F were among a panel of proinflammatory cytokines that could predict CKD progression [55]. In addition to other studies that have demonstrated that Th17 cells may contribute to pancreatic beta cell destruction [101], these observations suggest an important role for Th17 cells in diabetic nephropathy.
In streptozotocin-induced diabetes, mycophenolate treatment reduced Th17 cell infiltration into the kidney and alleviated the development of albuminuria and renal fibrosis without effects on glycemic control [102], while IL-17-/- mice, or mice treated with anti-IL-17 antibody were protected from the development of proteinuria and renal scarring [103]. Recently, Lavoz et al. [104] reported similar results; IL-17A immunoneutralization attenuated renal dysfunction and disease progression in BTBR ob/ob mice.
Renal inflammation also plays an important role in the development of hypertension [105,106]. Recent studies identified elevated levels of circulating IL-17 in patients with prehypertension, defined as a systolic BP of 120-139 [57]. In diabetic patients, IL-17 was elevated in those with hypertension vs. those without hypertension [56]. In another study of patients without blood pressure control, a correlation was observed between IL-17A level and the duration of hypertension [58].
Multiple animal models support the hypothesis that lymphocytes contribute to the development of hypertension. Genetic deletion of the Rag1 gene in Dahl salt-sensitive (S) rats resulted in depletion of mature T and B cells and a reduction in salt-sensitive hypertension [107]. In models of chronic angiotensin II (Ang II)-induced hypertension, the initial increase in blood pressure in response to Ang II infusion appeared largely independent of inflammation. However, sustained infusion of Ang II for up to 3 weeks in rats on a high-salt diet or in Dahl S rats resulted in increased renal lymphocyte accumulation, while mycophenolate treatment significantly abrogated the development of hypertension and renal fibrosis [108].
In C57BL/6J mice, Ang II infusion increased the percentage of circulating Th17 cells and IL-17A. IL-17A-/- mice showed similar elevations in blood pressure in the first 2 weeks of Ang II infusion as wild-type mice, but developed less severe hypertension (~30 mmHg) and reduced inflammation during the subsequent 2 weeks of infusion [56]. Similarly, Wade et al. [109] reported that treatment with sIL-17RC attenuated the degree of hypertension in Ang II-infused Dahl S rats. These data suggest that Th17 activation contributes to the full development of hypertension in Ang II-dependent models.
In other models, Amador et al. [110] demonstrated that mineralocorticoid-dependent hypertension was associated with activation of Th17 cells and the down-regulation of Foxp3 expression, while immuno-neutralization of IL-17 significantly attenuated the degree of hypertension and renal damage. Chiasson et al. demonstrated that the calcineurin inhibitor cyclosporine A increased expression of Th17 cells and decreased expression of Treg cells. The resultant increase in systolic blood pressure and impaired endothelial dependent vasodilation in isolated blood vessels was reversed by treatment with an anti-IL-17 antibody [111]. Finally, increased circulating levels of IL-17 have been demonstrated in patients with pre-eclampsia [59]. In a pregnant reduced uterine perfusion pressure (RUPP) rat model of pre-eclampsia, infusion of sIL-17RC significantly attenuated the development of hypertension [112].
It has been suggested that renal injury and antigen presentation promote activation of adaptive immune responses in the setting of hypertension or following acute injury (reviewed in [105,106,113]). Other physiological factors may influence the degree of lymphocyte activation and modulate Th17 responses. For example, increased dietary salt intake has been shown to increase Th17 expression not only in models of kidney injury, but also other models of Th17-dependent inflammation [114–117], suggesting that the effect of dietary salt is not dependent exclusively on kidney damage. In vitro studies demonstrated that elevated extracellular sodium concentration can hasten differentiation of naïve Th0 cells to a Th17 phenotype in vitro [118]. It has been suggested that a high-salt diet can increase Na+ deposition in extracellular glycosaminoglycans in the skin, which has been postulated to increase local Na concentration relative to extracellular fluid [119].
Our group demonstrated that Ang II in combination with elevated extracellular sodium (170 mM) synergistically enhanced IL-17 production in CD4+ lymphocytes isolated from post-ischemic kidneys [91]. In addition, losartan dramatically attenuated Th17 cell accumulation in response to a high-salt diet following ischemic AKI, suggesting that the Ang II pathway contributes directly to activation of these cells [91]. Recent studies have shown that patients with salt-losing tubulopathies have reduced skin sodium content associated with reduced Th17 activation and impaired immune responses. It was suggested that an altered ionic environment resulted in reduced Th17 cells activity as lymphocytes from these patients were able to activate the Th17 pathway in vitro in response to extracellular sodium [120]. These studies further indicate that sodium status has a direct impact on the activation of Th17 cells.
Effects of interleukin 17 effects on different renal cell types promote alterations in renal function and the development of fibrosis
The role of Th17 cells likely varies in different types of kidney disease due to underlying differences in the etiology of various kidney diseases and the highly pleiotropic nature of IL-17A. As described above, IL-17 receptors are localized in neutrophils, which contribute to the strong proinflammatory effect of Th17 cells in inflammatory conditions. However, the precise roles of Th17 cells in different kidney diseases remain to be fully elucidated. As part of this process, an appreciation of the different cell types with the potential to be influenced by IL-17A is required.
To date, few studies have directly evaluated the presence of IL-17 receptors in the various cell types of the kidney. However, gene profiling studies are providing insight into the dynamics of IL-17-IL-17R expression. For example, Liu et al. [121] utilized a transgenic RNA-translating ribosome affinity purification (TRAP) approach to profile genes in a cell-type specific fashion in response to I/R injury. By incorporating Cre-specific promoters to drive tagged ribosomal protein expression, RNA isolated from tagged ribosomes was used for cell specific gene profiling. A search of the Gene Expression Omnibus (GEO) public data set utilized in this study [121] indicated that the highest baseline (i.e., non-injury) expression of IL-17RA mRNA was in the epithelial compartment. However, following renal I/R injury, relative IL-17RA mRNA expression was significantly induced by ~2.5, ~4, and ~6-fold in interstitial, endothelial, and myeloid-derived cells, respectively. Therefore, in addition to its effects on neutrophils, the expression of IL-17 receptors on multiple cell types points to a complex role for IL-17 in renal pathophysiology (Fig. 2).

Pleiotropic effects of T helper 17 (Th17) cells on the development of interstitial fibrosis and the development of hypertension.
Cytokines released from injured kidney tissue, such as C-C motif chemokine ligand 20 (CCL20), may be chemoattractants for Th17 cells. Interleukin (IL) 17 release in the milieu of the kidney has the potential to influence fibrosis and hypertension by interacting with a number of cell types. IL-17 receptors are present on hematopoietic, epithelial, endothelial, and smooth muscle cells, as well as pericytes and fibroblasts. In tubules, IL-17 can stimulate activities associated with inflammatory cell recruitment, matrix accumulation, and enhanced sodium reabsorption. Pericytes or smooth muscle cells may acquire a more myofibroblastic phenotype. Evidence from endothelial cells and smooth muscle cells suggests that IL-17 may alter the regulation of vascular tone. Finally, IL-17 may have stimulatory or inhibitory effects on the inflammatory activation of macrophages and neutrophils depending on timing and context.
ECM, extracellular matrix; TGF, transforming growth factor; ROS, reactive oxygen species; NOS, nitric oxide synthase.
Effects on epithelial cells
IL-17 likely mediates blood pressure responses in part by regulating renal sodium transporters in the epithelium. Norlander et al. [122] demonstrated that IL-17A upregulation secondary to angiotensin infusion increased the expression of the sodium hydrogen exchanger-3 (NHE 3) and the sodium chloride cotransporter (NCC). IL-17A treatment of mouse distal convoluted tubules or human proximal tubule cells in vitro also increased expression of these sodium transporters, which was dependent on the phosphorylation of serine/threonine-protein kinase (SGK)-1 [122]. In vivo, mice exhibited impaired diuresis and natriuresis responses to acute saline loads following chronic Ang II infusion, and this was significantly attenuated in IL-17A-/- mice [123]. Damaged tubules also play an important role in the activation of inflammatory pathways leading to renal fibrosis [124,125], and IL-17 activity may contribute to this effect. In vitro studies of proximal tubule cells demonstrated that IL-17 stimulation increased the expression of profibrotic molecules and the production of IL-6 and IL-8, in part via activation of extracellular signal-regulated kinase 1/2 phosphorylation [50,126]. It is unclear if elevated IL-17 is sufficient to induce elevated blood pressure responses or if it serves to modulate other sodium-retaining factors to effect hypertension.
Effects on endothelial cells and vascular smooth muscle cells
Immune cells in the kidney may represent a source of oxidant stress. IL-17 has been shown to impair endothelial nitric oxide synthase activity, suggesting that altered vascular activity may also contribute to the development of hypertension [114,127]. A recent study by Orejudo et al. [128] suggested that vascular remodeling of small arteries is influenced by IL-17A. In this study, IL-17 induced hypertrophy of vascular smooth muscle cells in vitro, while IL-17 administration in mice increased blood pressure and induced inward remodeling of small mesenteric vessels. Hydralazine normalized blood pressure but had no effect on the inward remodeling induced by IL-17 [128]. IL-17 also stimulates endothelial cells to promote neutrophil recruitment by activation of p38MAPK and STAT3 [129,130]. In brain-derived endothelial cells, TNF-α and IL-17 increased the production of CCL2 and CXCL1; other cytokines including CCL20 and IL-17 facilitated the migration of Th17 cells through the endothelial monolayer [131].
Activation of smooth muscle-like pericytes is considered to play a central role in the development of interstitial fibrosis [132]. No studies have directly evaluated the effects of IL-17 on renal pericytes or myofibroblasts in the setting of kidney disease. However, Th17 cells play a key role in asthma, and IL-17 has been shown to direct remodeling of airway smooth muscle [133]. Studies of pericytes and smooth muscle cells from other organs suggest that these cells are highly responsive to IL-17. Gene array analysis of human aortic smooth muscle cells found that over 30 genes, including inflammatory cytokines and chemokines, were stimulated by incubation with IL-17 [56]. Cultured dermal or placental microvascular pericytes stimulated with IL-17 produced soluble factors that enhanced neutrophil migration and stabilized pericyte/neutrophil interactions [134,135].
Effects on macrophages
In a model of wound healing, IL-17 administration increased inflammation, aggravated fibrogenic scar formation, and delayed wound healing. Blockade of macrophages with clodronate reduced the inflammatory response to IL-17 and improved wound healing [136]. Ge et al. [137] investigated the effects of macrophage-specific deletion of IL-17RA in a model of UUO and demonstrated reduced accumulation of monocytes and renal fibrosis. It is unclear whether IL-17 influences macrophage polarization. One study reported that IL-17 stimulation of human monocytes did not alter M1/M2 markers but rather increased the expression of co-stimulatory molecules and proinflammatory cytokines in response to stimulation with oxidized low-density lipoprotein [138]. However, other studies suggested that IL-17 may mediate M2 polarization, consistent with an anti-inflammatory/pro-repair or fibrotic phenotype [139,140].
Beneficial effects of interleukin 17 on outcomes in kidney disease models
While the majority of evidence suggests that inappropriate activation of Th17 cells enhances inflammation and contributes to acute and CKD, these data should be viewed with caution, since several studies have reported that IL-17 may be protective in renal disease. Inhibition of IL-17 was reported to aggravate renal injury and fibrosis in a UUO model [141] and response to DOCA-salt/Ang II-induced hypertension [142]. An in vitro study demonstrated that IL-17 inhibited TGF-β stimulation of renal fibroblasts from IL-17-/- mice following UUO [143]. Overexpression of IL-17 by adenovirus reduced inflammation and renal fibrosis, which was suggested to be the result of the local kallikrein-kinin system altering the balance of extracellular matrix-degrading enzymes [96]. In C57BL6/lpr mice, a model of SLE, mutation of IL-17 blocked development of the SLE phenotype, but resulted in a significant lymphoproliferative disease characterized by splenomegaly and the expansion of total and double-negative T cells [144].
Mohamed et al. [145] showed that IL-17A levels in patients were reduced in advanced stages of diabetic kidney disease. In a streptozotocin model, IL-17A deficiency resulted in more severe disease progression relative to wild-type mice, and administration of low dose IL-17A reduced macrophage infiltration and the development of renal fibrosis. In our laboratory, RORγt mutant rats showed reduced sensitivity to AKI following I/R vs. wild-types rats. However, an increase in ischemic time in RORγt mutants resulted in failure to recover from AKI [86]. Administration of IL-17A facilitated recovery of postischemic RORγt mutant rats to a similar state as wild-type controls. Recovery from AKI due to IL-17 treatment was associated with inhibition of M1 macrophage infiltration, which remained elevated in untreated RORγT mutant rats for up to 4 days following I/R [86]. Taken together, these data suggest that the balance of Th17 cells plays an important role in immune homeostasis and may play an unappreciated role in feedback to suppress inflammation under some experimental conditions.
Concluding remarks
Since the discovery of Th17 cells, an important contributory role for these cells in kidney injury and fibrosis has been identified. We reviewed the accumulated research data implicating Th17 activation in models of kidney disease and hypertension. In addition, we outlined evidence that the primary cytokine produced by Th17 cells, IL-17A, may interact with multiple inflammatory and resident cells to drive disease. Due to its pleiotropic effects on multiple target cell types (outlined in Fig. 2), IL-17 is an attractive therapeutic target in several kidney disorders. Several reviews have addressed potential strategies to target IL-17 in disease, and biologic agents are already in use for treatment of diseases such as psoriasis [146]. An addition to biologics, other agents directed at SOCCs, which influence Th17 activation, are under development [147] and it is conceivable that such interventions may have therapeutic efficacy in patients with kidney disorders.
Notes
Conflict of interest
All authors have no conflicts of interest to declare.
Funding
Work from the authors presented in this paper was supported by National Institutes of Health grant DK-063114 and a Bridge Funding award from the Indiana University Research Foundation (DPB). Purvi Mehrotra was supported by an Indiana University Showalter Fellowship.
Authors’ contributions
Conceptualization: All authors
Writing–original draft: All authors
Writing–review & editing: All authors
All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Dr. Robert Bacallao for critical review of this manuscript and Ms. Barbara Sturonas-Brown for assistance with illustrations.