



Introduction
The prevalence of autosomal dominant polycystic kidney disease (ADPKD) is an estimated 12.5 million cases worldwide across all ethnicities [1]. ADPKD is the most common inherited kidney disease, accounting for 5% to 10% of global cases of end-stage kidney disease (ESKD) [1,2]. It is a systemic disease characterized by early development of fluid-filled renal cysts that relentlessly grow with time, leading to destruction of kidney parenchyma and loss of kidney function by the fifth to sixth decade of life [3]. There are about 0.6 million to 0.7 million cases of ADPKD in the United States [2,4]. Over the last decade, there have been major advancements in understanding the pathogenesis and natural history of ADPKD, including identification of several disease-causing mutations in genetically unresolved cases and development of disease-modifying treatment options such as tolvaptan. This review highlights the genetic mutations associated with ADPKD, defines patients at risk of rapid progression to ESKD, and focuses on the management of ADPKD in the era of disease-modifying agents.
Genetic variability in autosomal dominant polycystic kidney disease
ADPKD is a genetically heterogeneous disease that is inherited in an autosomal dominant manner [4]. In the majority of cases, it is attributed to mutations in either the PKD1 gene on chromosome 16 encoding polycystin (PC)-1 or PKD2 on chromosome 4 encoding PC-2, with the former responsible for 78% and latter about 15% of the cases [5]. There is wide phenotypic variability, with PKD1 mutations manifesting more severe disease including more numerous cysts, larger height-adjusted total kidney volume (TKV), lower estimated glomerular filtration rate (eGFR), and earlier development of ESKD compared to PKD2 mutations [4,5]. In addition to the genotype, several other factors contribute to the phenotypic variability observed in ADPKD. These factors include mosaicism, rate of cystic growth, and environmental influences such as water intake, diet, hormonal factors, obesity, and smoking [6,7].
Other diseases can present with kidney cysts and might mimic ADPKD. Thus, it is essential to understand these nuances to allow an accurate diagnosis, which will affect the prognosis and treatment plan. Mutations in PRKCSH, SEC63, LPR5, ALG8, and SEC61B that are associated with autosomal dominant polycystic liver disease (ADPLD) can result in renal cysts and an ADPKD-like phenotype without increased risk of progression to ESKD [8]. Recently, mutations in GANAB, which encodes the glucosidase II subunit α protein necessary for maturation of PC-1 protein, were found to cause mild kidney cystic disease (average 10 cysts total), mild decline in kidney function, and mild to severe polycystic liver disease. GANAB-associated disease represents 0.3% of patients with ADPKD [5,9]. Mutations in PKHD1, which are associated with autosomal recessive polycystic kidney disease (ARPKD) and present with congenital hepatic fibrosis, can mimic ADPKD [8]. Furthermore, mutations in UMOD, REN, MUC1, and HNF1B associated with autosomal dominant tubulointerstitial kidney disease (ADTKD) can present with renal cysts and low kidney function that can mimic ADPKD [8]. In contrast to ADPKD, patients with ADTKD present with smaller cystic burden (i.e., normal to mildly enlarged kidneys and relatively small number of kidney cysts). The predominant feature in ADTKD is interstitial fibrosis, which leads to progressive loss of kidney function [10]. In a recent study by Cornec-Le Gall et al. [11], mutations in DNAJB11 were found to be associated with an ADPKD-like phenotype with an overlap of ADTKD clinical characteristics and the presence of liver cysts. Additional mutations that cause kidney and liver cysts without enlargement of kidneys include those in ALG9 [12].
Mutations associated with impaired ciliary apparatus function and ciliopathies such as those found in OFD1 and NPHP1 can present with corticomedullary cysts in the kidneys that might mimic ADPKD but with distinct extrarenal manifestations and ESKD onset at a younger age [13,14]. Other systemic syndromes such as tuberous sclerosis complex and Von Hippel Lindau disease, due to mutations in TSC and VHL genes, respectively, can present with kidney cysts mimicking ADPKD [15]. Hence, there is considerable phenotypic overlap between ADPKD and other inherited cystic kidney diseases, highlighting the importance of accurate diagnosis of ADPKD as it will affect the renal prognosis and treatment plan to slow the disease process.
Diagnosis of autosomal dominant polycystic kidney disease and situations when genetic testing is required
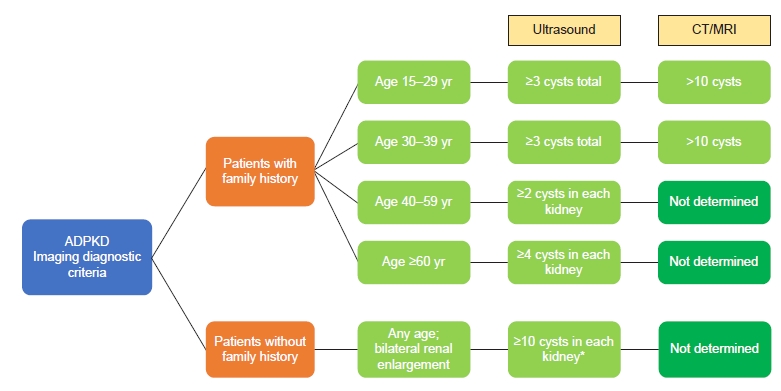
In most cases, ADPKD is diagnosed clinically by evaluating the number of kidney cysts on imaging adjusted to age in the presence of family history of ADPKD [16]. Fig. 1 summarizes the diagnostic criteria for both ultrasound and computed tomography (CT)/magnetic resonance imaging (MRI) in the presence or absence of family history. In the absence of family history, there are no established criteria. Expert opinion suggests that bilateral renal enlargement with innumerable renal cysts (>10 cysts per kidney) provides a “likely ADPKD” diagnosis [4]. In these situations, molecular genetic testing would be prudent to confirm the diagnosis. As genetic screening is becoming more readily available, the indications for testing will likely be more inclusive as these results enrich the prognostication in ADPKD. Until genetic testing becomes universally accessible, the following indications are considered: 1) confirm the ADPKD diagnosis in the setting of negative family history, 2) ascertain the diagnosis if the extrarenal manifestations are suggestive of syndromes other than ADPKD or if the cystic burden is not congruent with the renal function, 3) exclude ADPKD in young potential kidney donors who are at risk of ADPKD, and 4) confirm the diagnosis and rule out ciliopathies in the setting of early or very early disease onset [17]. Fig. 2 summarizes the indications for genetic testing in patients with renal cysts suspicious for ADPKD.
Assessment of risk of rapid progression in autosomal dominant polycystic kidney disease
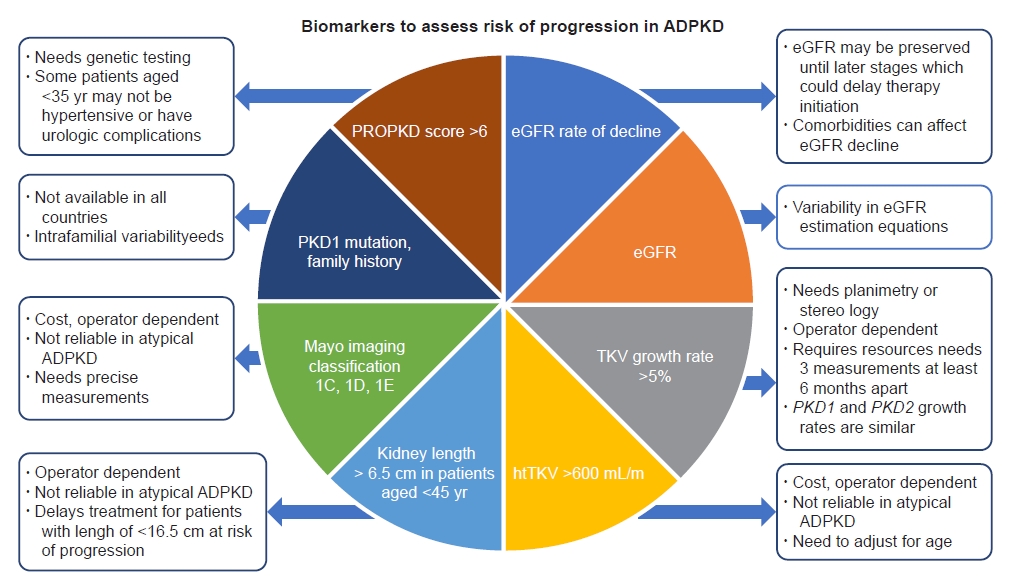
Not all patients with ADPKD reach kidney failure. In fact, 50% of the patients reached ESKD by age of 54 years and 75% by age of 62 years in a recent study with a large tertiary care center cohort [18]. It has also been demonstrated that, within a family, despite shared mutations, there is wide variability in the age at which a family member reaches ESKD [19]. Given the wide heterogeneity and phenotypic variability of ADPKD, it is prudent to identify the group of patients who are at risk of rapid progression toward ESKD to initiate disease-modifying treatment early to slow the progression of the kidney disease. There are several methods available with variable advantages and disadvantages to assess the risk of progression in ADPKD (Fig. 3). Irrespective of the method, the most important factor in this assessment is matching the cystic burden and kidney function with respect to age, as following thresholds indiscriminately might lead to misclassification of the disease and its progression.
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) is an ongoing, prospective, multicenter, observational study of 241 ADPKD patients (aged 15 to 45 years with creatinine clearance of >70 mL/min at the time of study initiation) that determined the relationship between kidney volume and decline in kidney function using MRI [20]. Key findings from the study include that exponential increase in TKV is variable among patients, TKV is related to decline in glomerular filtration rate (GFR), and baseline height-adjusted TKV (htTKV) predicts decline of kidney function and progression to kidney failure, validating it as a prognostic marker in ADPKD [21,22]. If there is a discordance between htTKV, age, and decline in kidney function, an alternative diagnosis such as ADTKD and other mimickers of ADPKD should be considered as described above. When interpreting TKV, it is prudent to adjust to the age at time of imaging and ensure that ADPKD is typical in its radiological appearance. These two critical factors are embedded in the Mayo imaging classification (MIC) [23]. This classification delineates the criteria for typical (class 1 or MIC 1) and atypical (class 2 or MIC 2) ADPKD. MIC 1 is defined as bilateral and diffuse renal cystic disease where cysts are contributing almost equally to TKV. MIC 2 is defined as either unilateral, focal, asymmetric cystic involvement (MIC 2A) or bilateral cystic involvement with kidney atrophy (MIC 2B). Furthermore, MIC 1 is divided into five subcategories (MIC 1A through 1E) that predict the rate of kidney volume growth by adjusting htTKV to age. Patients with MIC 1C, 1D, or 1E are considered at risk of rapid progression as their future eGFR decline is predicted to be ≥2.5 mL/min/1.73 m2 per year and the mean age of ESKD onset is ≤54 years. The MIC has been validated in a study by Yu et al., and it was concluded that htTKV is a long-term predictor of decline in GFR [24]. In a study by Bae et al. [25], htTKV was recalculated after exclusion of prominent exophytic cysts, and the imaging classification based on recalculated htTKV was more predictive of decline in eGFR. The definition of rapid progression is evolving. There is currently controversy on treating MIC 1C, where the United States practical guide favors treating with tolvaptan, whereas the updated recommendations from the European Renal Association-European Dialysis and Transplantation Association and PKD International advise seeking additional confirmatory evidence of rapid progression such as age-adjusted GFR, genotyping, predicting renal outcome in ADPKD (PROPKD) score, or family history [26]. While there is no consensus, international guidelines will likely be developed through an upcoming Kidney Disease: Improving Global Outcomes (KDIGO) workgroup.
The gold standard for measurement of TKV is planimetry or stereology in which kidney volume is calculated by tracing the kidney in cross-sectional slices or coronal slices with corresponding slice thickness. However, this method is time-consuming and requires specialized training and equipment [27]. A practical alternative includes TKV measurement using an ellipsoid formula that measures sagittal and coronal length, width, and depth. Estimating TKV by ellipsoid equation has been demonstrated to be comparable to planimetry and provides TKV in a few minutes [23,27]. A model developed by the Mayo Clinic for calculating TKV by the ellipsoid formula allows classification of patients into MIC 1A through E and predicts future eGFR decline (http://www.mayo.edu/research/documents/pkdcenter-adpkd-classification/doc-20094754).
Experts across the globe have used various biomarkers to assess the risk of ADPKD progression. Fig. 3 summarizes the various prognostic biomarkers for predicting the risk of rapid progression. The biomarkers include clinical, genomic, and radiological criteria to assess the rate of kidney growth and kidney function decline. Annual TKV growth rate of >5% measured by planimetry or stereology has been considered a radiologic biomarker for risk of rapid progression in Japan. However, its applicability is limited given the need for precise measurements and inability to differentiate between PKD1 and PKD2 due to similar TKV growth in these patients [18]. Use of kidney length as a biomarker can delay treatment initiation for young patients with kidney size smaller than 16.5 cm who might be at risk of rapid progression to ESKD [18]. Conversely, the use of kidney length alone might misclassify patients with atypical ADPKD (focal disease with few large kidney cysts) as rapid progressors. Genomic and clinical data such as PKD1 vs. PKD2 mutation status (truncating vs. non-truncating) and family history of developing kidney failure at a younger age might predict the severity but are highly variable at population as well as intrafamilial levels [5–7]. Thus, these factors would be helpful to enrich prognostication but lack the individual precision for patients when used in silico without the overall clinical context. The PROPKD scoring system incorporates sex, onset of hypertension before age of 35 years, urologic events before age of 35 years, and type of mutation (PKD1 truncating, PKD1 non-truncating, and PKD2) to predict the risk of rapid progression to ESKD before age of 60 years. This system has a positive predictive value of 91% for a score of >6 but is limited by cost and availability of genetic testing and lacks accuracy due to phenotypic variability within a family [28]. European regulatory agencies have used eGFR rate of decline of at least 2.5 mL/min/1.73 m2 per year over 5 years or at least 5 mL/min/1.73 m2 in 1 year as a marker for rapid progression. However, use of this marker alone can delay treatment initiation in younger age groups at risk of rapid progression but with preserved kidney function and might erroneously include patients who have concomitant diagnoses that lead to GFR decline independent of ADPKD risk of progression such as diabetic nephropathy [18]. In our opinion, the most practical approach available at the moment is the MIC system, which allows prediction of the intrinsic TKV rate of growth using one htTKV measurement adjusted by age. Patients with MIC 1C, 1D, or 1E are considered at risk of rapid progression.
Basic kidney protective measures in autosomal dominant polycystic kidney disease
Basic kidney protective measures such as control of blood pressure, limiting dietary sodium and caloric intake, hydration, and management of dyslipidemia must be implemented in all ADPKD patients irrespective of progression risk. Blood pressure in ADPKD patients aged 18 to 50 years who are at risk of rapid progression (MIC 1C, 1D, or 1E) should be targeted to less than 110/75 mmHg as this strict blood pressure control could slow the TKV rate of growth and potentially slow the decline in eGFR [29,30]. In all other ADPKD patients, blood pressure can be targeted to less than 130/80 mmHg [29]. Renin-angiotensin-aldosterone blockade with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are the recommended first-line therapy for management of hypertension in ADPKD patients [29]. In addition, patients should be on moderate dietary sodium restriction (2.3–3.0 g) and daily dietary protein intake of 0.8–1.0 g/kg ideal body weight to slow progression of chronic kidney disease [29]. Based on animal model observations, enhanced water intake to suppress vasopressin levels might be associated with slower TKV growth and eGFR decline. Hence, all ADPKD patients should be advised to increase hydration to target a urine osmolality of <280 mOsm/L [29]. In a recent multicenter, open-label, randomized control clinical trial studying the effect of a water prescription on slowing the disease process in ADPKD, the prescribed water prescription was not associated with slower growth of TKV over 3 years compared with ad libitum water intake. However, only 50% of the patients were able to reach the target urine osmolality despite coaching, indicating that maintaining a suppressed vasopressin level without pharmacological intervention is challenging in a practical clinical setting. Additionally, 30% of the patients included in this clinical trial were at risk of slow progression (MIC 1B), which could have affected the study result as interventions are less likely to be shown in a short period of time in this group given their small TKV rate of growth [31,32]. Other recommendations include treatment of dyslipidemia to target low-density lipoprotein (<100 mg/dL) and high-density lipoprotein (HDL, >50 mg/dL) and maintaining normal body mass index (BMI) as lower serum HDL cholesterol and higher BMI have been associated with faster increase in TKV and decline in GFR [29,33].
Vasopressin antagonists in autosomal dominant polycystic kidney disease
In 2018, the U.S. Food and Drug Administration approved tolvaptan, a vasopressin V2 receptor antagonist, as a disease-modifying treatment option for ADPKD patients at risk of rapid progression to kidney failure [34]. The PC proteins present across the cilium in kidney tubules regulate intracellular calcium and cyclic adenosine monophosphate levels. Mutations of these proteins lead to elevation in cyclic adenosine monophosphate (cAMP) level and eventual dysregulation of several downstream pathways leading to cyst formation, fluid secretion, interstitial inflammation, and fibrosis [8]. Vasopressin has been demonstrated to reduce cAMP level in mouse models through its antagonism of the V2 receptor and subsequently inhibition of cystogenesis [35].
In 2012, a randomized, double-blind, placebo-controlled trial (TEMPO 3:4) was conducted to evaluate tolvaptan efficacy in ADPKD patients aged 18 to 50 years with creatinine clearance greater than 60 mL/min and TKV of >750 mL [36]. Tolvaptan treatment was associated with a slower rate of increase in TKV (2.8% vs. 5.5%, p < 0.001) as well as a slower decline in kidney function in the tolvaptan group compared to placebo [36]. This was followed by another randomized, double-blind, placebo-controlled clinical trial (REPRISE) in 2017 to evaluate the efficacy of tolvaptan in ADPKD patients aged 18 to 65 years with eGFR of 25 to 65 mL/min [37]. The results were consistent with the previous study, with a slower decline in eGFR in the tolvaptan group compared to placebo (2.34 mL/min/yr vs. 3.61 mL/min/yr, p < 0.001) [37]. Tolvaptan was associated with a 5.6% risk of hepatic aminotransferase elevation compared to 1.2% in the placebo arm. This hepatic enzyme dysregulation was reversible after discontinuation of the drug [37].
Tolvaptan is currently recommended for ADPKD patients in the United States aged 18 to 55 years, at risk of rapid progression as determined by htTKV and risk of rapid GFR decline in the future (MIC 1C, 1D, 1E) with an eGFR greater than 25 mL/min [18]. For patients aged 55 to 62 years, emerging evidence favors the use of tolvaptan. Shared decision-making is recommended in this age group for patients who have evidence of rapid progression (i.e., MIC 1C, 1D, or 1E with an average eGFR rate of decline ≥ 2.5 mL/min/yr over the past 5 years). The shared decision-making will entail a discussion of risks, potential benefits, and patient preferences. Fig. 4 depicts examples of patients who should and should not be considered for disease-modifying treatment options. Patients with discordance between htTKV and eGFR should be evaluated for concomitant disease processes such as diabetic nephropathy, vascular disease, or ADPKD mimickers such as ADTKD and should not be considered for disease-modifying treatment options. Representative cases covering common clinical scenarios are detailed in the section below.
Most common adverse effects of tolvaptan resulting from vasopressin receptor blockade that patients should be advised of are polyuria, excessive thirst, polydipsia, and nocturia [38]. Idiosyncratic hepatocellular injury in the form of elevated aminotransferases was observed in 4.4% and 5.6% of patients treated with tolvaptan in the TEMPO 3:4 and REPRISE trials, respectively. A risk evaluation and mitigation strategy with frequent liver function monitoring before and after initiation of tolvaptan are required to assess abnormalities in aminotransferases and risk of serious hepatoxicity that might necessitate discontinuation of tolvaptan [38].
Representative cases
Once ADPKD diagnosis is confirmed, the next step involves assessing the risk of rapid progression. Fig. 4 summarizes the various clinical scenarios where disease-modifying treatments such as tolvaptan are indicated or not and where shared decision-making is required to balance risks and benefits with patient preference.
Case 1
A 29-year-old female with typical ADPKD (MIC 1) had a TKV of 2,627 mL, corresponding to MIC 1E. Her eGFR was already low at 29 mL/min/1.73 m2 despite her young age. Thus, she was considered at risk of rapid progression and was showing evidence of rapid progression given the low GFR. However, her kidney length was only 16 cm, which highlighted the disadvantage of only using this criterion to assess rapid progression. Furthermore, her PKD mutation type (PKD1 non-truncating) and PROPKD score of 3 had a good relative prognostication. This highlights the lack of individualized prognosis when only using genomic data and highlights the importance of not using these factors in silo to assess rapid progression. Tolvaptan could be continued until the need for renal replacement therapy.
Case 2
A 30-year-old male with typical ADPKD (MIC 1) had a TKV of 1,351 mL and MIC 1D. His eGFR was 81 mL/min/1.73 m2. Despite his PKD1 truncating mutation, his prognosis was better than that of case 1 (with PKD1 non-truncating mutation), highlighting the complex interaction of factors that dictate disease progression beyond the PKD genotype. This patient would benefit from tolvaptan as he was at risk of rapid progression based on the MIC.
Case 3
A 35-year-old male with typical ADPKD, TKV of 820 mL, MIC 1C, and eGFR of 85 mL/min/1.73 m2 was eligible for tolvaptan treatment as he is at risk of rapid progression despite his favorable PROPKD score and PKD2 non-truncating mutation.
Case 4
A 52-year-old female with typical ADPKD, TKV of 380 mL, MIC 1A, and eGFR of 76 mL/min/1.73 m2 was not eligible for tolvaptan as she is considered a slow progressor despite a PKD1 non-truncating mutation.
Case 5
A 51-year-old female with typical ADPKD, TKV of 1,131 mL, MIC 1B, and eGFR of 51 mL/min/1.73 m2 who would not benefit from tolvaptan given the risk outweighing the benefits. Despite the patient carrying the PKD1 truncating mutation, her prognosis was very good. This case highlights the complex interaction with other factors that determine disease progression. These factors can include other genetic factors such as modifiers and epigenetic modifications and environmental factors such as healthy lifestyle including hydration, normal BMI, and dietary restrictions.
Case 6
A 46-year-old male with atypical ADPKD (MIC 2A, lopsided). TKV was high at 2,760 mL. However, this patient had atypical features including four very large cysts that accounted for >50% of the TKV. Correlating TKV with cystic burden and renal parenchyma as well as kidney function was essential to assess prognostication. This patient was predicted to have a good prognosis (slow progressor) and was less likely to reach ESKD at an early age. In this patient, kidney length was large, which would have misclassified the patient as a rapid progressor if the cystic burden was not evaluated on CT/MRI using the imaging classification system.
Case 7
A 53-year-old female with typical ADPKD, TKV of 517 mL, and MIC 1A had a low eGFR of 26 mL/min/1.73 m2 discordant with her low renal cystic burden, raising the question of a possible concomitant process leading to such low GFR. Additional investigation revealed oxalate nephropathy. Thus, the GFR decline was not related to ADPKD, and disease-modifying treatments directed to slow cystic disease progress were not indicated. This case highlights the importance of evaluating GFR rate of decline and chronic kidney disease stage in the context of cystic burden.
Case 8
A 65-year-old male with atypical ADPKD, TKV of 619 mL, and MIC 2B (bilateral renal cysts with low eGFR and atrophic kidneys). His low GFR was consistent with MIC 2B, which carries a poor prognosis as the main process is renal atrophy and renovascular disease in the setting of vascular pathology. Disease-modifying treatments directed to slow cystic disease progress were not indicated.
Case 9
A 67-year-old male with bilateral renal cystic disease. The relatively low cystic burden (TKV of 561 mL) and low eGFR (27 mL/min/1.73 m2) were discordant, raising the question of a renal cystic disease other than ADPKD or ADPKD with another concomitant renal disease. Thus, genetic study was indicated. This patient had DNJAB11-associated disease, which also has autosomal dominant inheritance with interstitial fibrosis as a major contributor to GFR decline.
Case 10
A 57-year-old male with typical ADPKD, TKV of 3,558 mL, and MIC 1D. His eGFR was 39 mL/min/1.73 m2. This patient had evidence of rapid progression and would benefit from initiation of tolvaptan to slow his disease process. However, there should be discussion with the patient for shared decision-making to balance risks, benefits, and patient preferences.
Conclusion
In the era of disease-modifying treatments intended to slow the disease progression of ADPKD, five main points need to be addressed in an individualized fashion: 1) confirm the diagnosis of ADPKD by ensuring the cystic burden matches the observed kidney function; 2) assess the risk of rapid progression using available biomarkers such as age and htTKV; 3) implement renal protective measures for all ADPKD patients; 4) evaluate eligibility for disease-modifying treatments such as tolvaptan by discussing the risks, benefits, and patient preference; and 5) implement safe prescription of tolvaptan based on regulatory guidance for serial liver function testing.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









