



Introduction
The nephron segment, cortical thick ascending limb (cTAL) regulates sodium chloride (NaCl) reabsorption as one of the most involved segments in NaCl retention states. In normal conditions, absorption of NaCl is balanced with its excretion, maintaining Na balance. This homeostasis is the final result of the action of molecules increasing and decreasing transport. However, in pathologic conditions, such as congestive heart failure, chronic kidney disease, hypertension, cirrhosis, and other diseases, Na retention is greater than its excretion, leading to a positive Na imbalance. This Na overload occurs because Na retention factors overcome the actions of molecules that increase its excretion, resulting in resistance to natriuretic effects. Understanding the interaction of the autacoids at the different nephron segments, especially at the cTAL, would help to elucidate such deleterious conditions and find strategies to manage them.
Previously, we reported that 8-iso-prostaglandin-F2╬▒ (8-iso-PGF2╬▒) increases Na+ and ClŌĆō reabsorption at the cTAL [1]. This effect, in vivo, would increase Na balance, inducing a blood pressure elevation that could be displayed as hypertension, congestive heart failure, cirrhosis, chronic kidney disease, or other edematous states. On the other hand, nitric oxide (NO) inhibits Na+ reabsorption at the cTAL, and this effect, in vivo, would induce diuresis and natriuresis, a mechanism that would decrease the described Na gain [2,3]. Despite the clear understanding of the importance of these effects, the interaction of NO and 8-iso-PGF2╬▒ in the cTAL has not been evaluated to elucidate their outcomes.
Na+ regulation has been previously reported at several nephron segments mediated by agents such as NO and angiotensin II in the cTAL [4,5], NO and arginine vasopressin at the cortical collecting duct level [6], and other segments [7]. The final effect of the interaction between NO and 8-iso-PGF2╬▒ is based on the intracellular activation of different regulators. Actually, whereas NO inhibits NaCl reabsorption by increasing intracellular cyclic guanosine monophosphate (cGMP) and decreasing cyclic adenosine monophosphate (cAMP) levels [7], 8-iso-PGF2╬▒ increases cAMP. The final effect will be elicited when one cyclic nucleotide preponderantly represses the other. When attempting to understand the crosstalk regulation of cyclic nucleotide concentrations, one must consider that such regulation can occur at the level of cyclic nucleotide synthesis, phosphodiesterase-mediated degradation, or even cellular efflux. Thus, we hypothesized that, in cTAL, persistent effects of 8-iso-PGF2╬▒ on Na reabsorption prevail over the shorter-acting NO effects, and protein kinase A (PKA) activation is required for such interaction.
Methods
Animals
Male New Zealand white rabbits (700ŌĆō1,200 g) were housed for 3 to 7 days before the study for acclimatization. The experiments were approved by the Institutional Animal Care and Use Committee of the J. Robert Cade Foundation (No. FJRC #2009) in C├│rdoba, Argentina. All animals were housed and handled according to the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Preparation and dissection of the cortical thick ascending limb of Henle
Animals were anesthetized with sodium thiopental (30 mg/kg, intravenously) and xylazine (20 mg/kg, intramuscularly) plus ketamine (50 mg/kg, intramuscularly). The kidney was exposed and bathed in ice-cold saline and then excised and cut into coronal slices along the longitudinal axis. The slices were placed in oxygenated physiological saline in a temperature-regulated chamber at 4┬░C. The cTAL of Henle was dissected from the medullary ray under a stereomicroscope as previously described [6,8].
Perfusion of thick ascending limbs
cTALs in a temperature-regulated chamber were perfused between concentric glass pipettes at 37┬░C as described [6]. The basolateral bath was exchanged at a rate of 0.5 mL/min. A total of five to eight tubules were analyzed during each protocol. Time-control experiments were conducted to ensure the stability of tubular transport. Chloride absorption (JCl) was measured as previously described [6,9]. cTALs were perfused and equilibrated for 20 minutes, and basal Cl absorption was calculated two to three times. Tested compounds, including 8-iso-PGF2╬▒, were either added to or removed from the lumen or bath as indicated. After a 20-minute equilibration period, tubular fluid was again collected 2 or 3 times.
Chloride concentrations in the perfusate and collected fluid
Chloride concentrations were measured by microfluorometry [10]. Because water is not reabsorbed by the thick ascending limb, JCl was calculated by the following equation: JCl = CR ├Ś (C0Cl ŌĆō C1Cl), where CR = collection rate normalized/tubule length, C0Cl = chloride concentration in the perfusion solution, and C1Cl = chloride concentration in the collected fluid.
Cyclic adenosine monophosphate
cAMP was measured as previously described [1]. Briefly, tubules were isolated and incubated in 95 ┬ĄL perfusion solution containing 1-mM 3-isobutyl-1-methylxanthine at 37┬░C for 10 minutes before adding 5-┬ĄL medium containing inhibitors or hormones. The reaction was stopped 30 minutes later with methanol (100 ┬ĄL), and cAMP and cGMP levels were measured by an enzyme immunoassay (EIA; Cayman Chemical, Ann Arbor, MI, USA). The samples were centrifuged, the supernatant was transferred to another tube and dried in a Savant centrifugal vacuum concentrator, and the pellet was reconstituted in 110-┬ĄL sodium acetate buffer. The cAMP standard was treated the same way. The results were expressed as fmol/min/mm tubule length.
Results
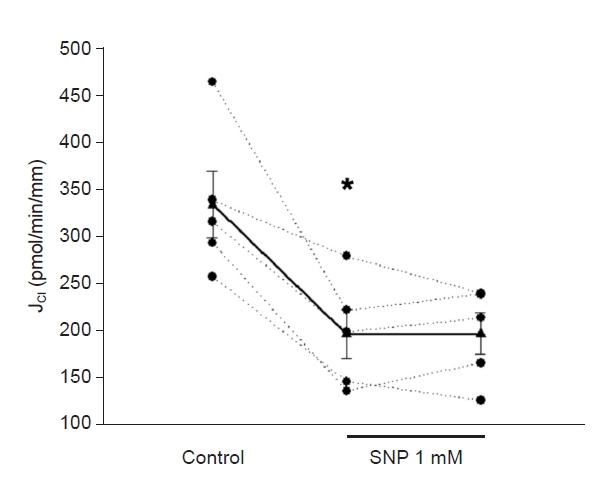
Since the 8-iso-PGF2╬▒/NO interaction in cTAL is uncertain, we chose a maximum NO bioavailability model. As described, the micro-isolated rabbit cTALs were transferred to a temperature-regulated chamber and perfused between concentric glass pipettes at 37┬░C [6] with the NO donor sodium nitroprusside (SNP) in a concentration that decreases transport by 40% as endogenous NO is inhibited in this nephron segment [11]. Before adding SNP to the bath, JCl was 333 ┬▒ 35 pmol/min/mm. SNP (1 mM) in the bath reduced JCl to 195 ┬▒ 26 pmol/min/mm (p = 0.01 vs. basal; n = 5) (Fig. 1), a 41% decrease, and remained inhibited for the rest of the experiment (196 ┬▒ 22 pmol/min/mm). In vivo, this reduction in JCl should increase diuresis. In time-control experiments, JCl remained constant throughout the experimental period.
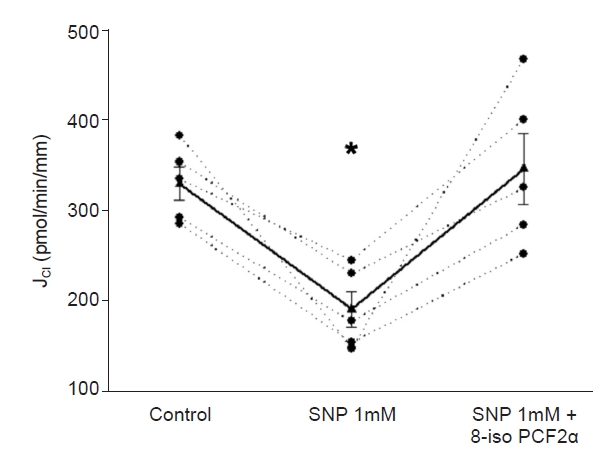
Next, we tested whether 8-iso-PGF2╬▒ stimulates JCl in the presence of SNP. Basal JCl was 330 ┬▒ 18 pmol/min/mm; when SNP (1 mM) was added to the bath, JCl decreased to 191 ┬▒ 19 pmol/min/mm. After adding 8-iso-PGF2╬▒ (100 ╬╝M) to the lumen, JCl increased to 346 ┬▒ 39 pmol/min/mm (p < 0.02 vs. NO; n = 5) (Fig. 2). We tested this concentration since those lower did not affect JCl [1]. Next, we evaluated the effect of 8-iso-PGF2╬▒ present in the basolateral side. Basal JCl was 235 ┬▒ 38 pmol/min/mm, which decreased in the presence of SNP (1 mM) to 139.4 ┬▒ 27 pmol/min/mm and increased to 297 ┬▒ 29 pmol/min/mm in the presence of 8-iso-PGF2╬▒ (100 ╬╝M; p < 0.02 vs. NO; n = 5) (Fig. 3). These data indicate that the maximal NO effect on cTAL is insufficient to prevent the 8-iso-PGF2╬▒-stimulated NaCl reabsorption.
In cTAL, 8-iso-PGF2╬▒ stimulates JCl via a cAMP-dependent mechanism [1], while NO inhibits chloride reabsorption by stimulation of a cGMP-stimulated phosphodiesterase, which decreases cAMP level [12]. Thus, we evaluated the effect of 8-iso-PGF2╬▒ on cAMP in the presence of a NO donor, SNP (1 mM). Basal cAMP was 56.3 ┬▒ 13.1 fmol/min/mm, while that in the presence of the NO donor was 57.8 ┬▒ 6.1 fmol/min/mm, the addition of 8-iso-PGF2╬▒ to which increased cAMP to 92.1 ┬▒ 2.9 fmol/min/mm (n = 10, p < 0.04) (Fig. 4). Thus, 8-iso-PGF2╬▒-stimulated NO-inhibited JCl is associated with a 60% increase in cAMP.
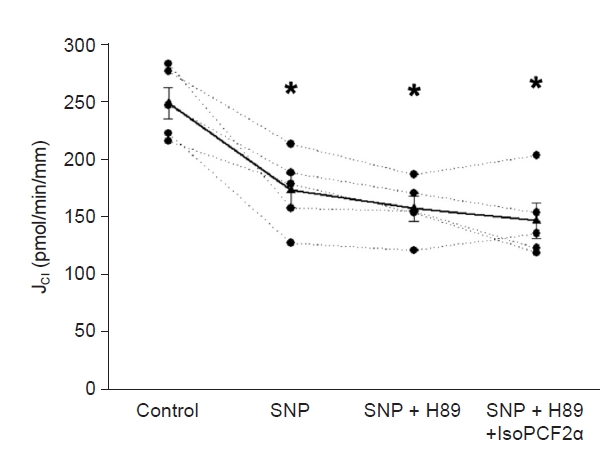
In the cTAL, PKA mediates the effect of 8-iso-PGF2╬▒ on JCl. Thus, we evaluated the effect of 8-iso-PGF2╬▒ on SNP-inhibited JCl in the presence of H89 (10 mM), a PKA inhibitor. Basal JCl was 249 ┬▒ 13 pmol/min/mm and decreased to 173 ┬▒ 14 pmol/min/mm when SNP (1 mM) was added to the bath. Addition of H89 to the bath did not change JCl significantly (158 ┬▒ 10 pmol/min/mm). Under these conditions, 8-iso-PGF2╬▒ could not stimulate SNP-inhibited JCl (147 ┬▒ 15 pmol/min/mm) (Fig. 5). Time control did not change during the experiment (280 ┬▒ 22 pmol/min/mm vs. 282 ┬▒ 36 pmol/min/mm). These data indicate that 8-iso-PGF2╬▒ requires PKA activity to reverse the NO-inhibited JCl.
Discussion
To our knowledge, this is the first report showing that 8-iso-PGF2╬▒ can override NOŌĆÖs diuretic effects in the cTAL, suggesting that Na+ retention will prevail over Na+ excretion in clinical conditions associated with increased 8-iso-PGF2╬▒ level. Although in the kidney, discrete 8-iso-PGF2╬▒ and NO effects have both been shown in vivo and in vitro, we designed this in vitro investigation to identify the local interaction and final effect of these two autacoids on chloride transport at the nephron segment.
Despite the well-known importance of prostaglandin in Na reabsorption, the effects of its derivatives are less clear. We evaluated the effects of 8-iso-PGF2╬▒ binding to the G-protein coupled receptor thromboxane A2 (TXA2) receptor, localized in the thick ascending limb segment [13,14]. This 8-iso-PGF2╬▒ is a modified prostaglandin produced by nonenzymatic oxidation of prostaglandin-F2╬▒ by reactive oxygen species [15,16] and induces vasoconstriction in the kidney and can impact renal blood flow [17]. Our data demonstrate that the NO donor decreases chloride reabsorption in the thick ascending limb of Henle; under these circumstances, 8-iso-PGF2╬▒ is activated by PKA and stimulates ClŌĆō reabsorption. In vivo, this interaction results in a decrease of natriuresis and sodium increase. The significance of this interaction resides not only in improving understanding of the normal physiology, but also in the prevention and treatment of many conditions such as hypertension, chronic renal failure, and cirrhosis, where isoprostanes increase and an Na retention state prevails [18].
The compound 8-iso-PGF2╬▒ binds the TXA2 receptor, a peroxidized derivative of prostaglandin-F2 localized in the basolateral and apical membrane of the thick ascending limb segment, which then activates cAMP production [14]. Following the release of 8-iso-PGF2╬▒, sudden activation of the TXA2 receptor in the thick ascending limb occurs [1], stimulating chloride reabsorption via cAMP. Simultaneously, NO inhibits 8-iso-PGF2╬▒ via cGMP generation. Therefore, the interaction between these two intracellular mechanisms could regulate sodium transport.
In the kidney, 8-iso-PGF2╬▒ induces vasoconstriction, which can modulate renal blood flow [19]. In the thick ascending limb, TXA2 receptor release stimulates PKA and induces an increase in chloride reabsorption across the Na-K-Cl cotransporter 2 (NKCC2) [20]. Increased concentrations of 8-iso-PGF2╬▒ and other isoprostanes have been observed in both the urine and plasma of hypertensive subjects. The enhanced NaCl retention in response to 8-iso-PGF2╬▒ might contribute to the pathogenesis of hypertension [20]. In the distal nephron, NO decreases cAMP, which reduces fluid absorption via PKA [21].
We further investigated whether 8-iso-PGF2╬▒ could reverse NO effects when PKA is inhibited. This experiment should establish whether the interaction between the mechanisms linking 8-iso-PGF2╬▒ and NO precedes or follows activation of PKA. In the presence of PKA inhibition, 8-iso-PGF2╬▒ did not change JCl, indicating that PKA activity is required to reverse the NO-inhibited JCl. Moreover, these results indicate that 8-iso-PGF2╬▒ reverses NO-induced inhibition of JCl via a mechanism involving activation of PKA. Since PKA is mainly activated by cAMP, we studied cAMP and found that NO was not able to reduce the cAMP level induced by 8-iso-PGF2╬▒.
Nevertheless, the effects of 8-iso-PGF2╬▒ and NO on NaCl excretion have been shown in several models [22]. The decreased urinary Na+ excretion caused by systemic nitric oxide synthase (NOS) inhibition correlates with higher urinary 8-iso-PGF2╬▒ [23,24]. Also, the superoxide scavenger Tempol blunts these changes, suggesting that inhibition of NO synthesis enhances endogenous superoxides, which could increase renal 8-iso-PGF2╬▒ level. These studies, however, show correlation but no direct interaction in the transport mechanisms. At any rate, 8-iso-PGF2╬▒ increases ClŌĆō by stimulating cAMP synthesis [1].
In contrast, the effect by which NO inhibits NKCC2 activity might involve both an increase in cGMP [12,25,26] and a decrease in cAMP [27]. In our study, the NO donor did not decrease cAMP, whereas cGMP can play an essential role in the NO-inhibited JCl. This effect might involve protein trafficking, limiting the total pool of NKCC2 at the membrane level [28] since 8-iso-PGF2╬▒ŌĆÖs increase of JCl is mediated by NKCC2 [1].
The mechanism(s) by which cGMP acts to regulate trafficking of the transporter might involve a decrease in insertion of the transporter into the membrane. Although evidence exists supporting the constitutive degradation of surface NKCC2, which is hypothesized to occur by decreasing NKCC2 level due to increasing NKCC2 ubiquitination and proteasomal degradation [28], it appears plausible that NO acts by increasing cGMP, which decreases cAMP [27] to lead to a reduction in exocytic insertion of NKCC2 into the apical membrane, reducing the number of transporters [19]. This process results in lower NKCC2 activity and blunted net NaCl reabsorption.
Our experiments demonstrate that interaction with 8-iso-PGF2╬▒ increases intracellular cAMP; in the presence of H89, 8-iso-PGF2╬▒ does not increase chloride reabsorption. These results show that the interaction occurs after activation of PKA since H89 prevented 8-iso-PGF2╬▒ and JCl inhibited NO and not because 8-iso-PGF2╬▒ blunts cGMP formation. Indeed, we found that NO caused no significant change in 8-iso-PGF2╬▒-stimulated cAMP level. This indicates that NO-stimulated phosphodiesterase activity has little or no effect on 8-iso-PGF2╬▒-stimulated cAMP level. This notion might not apply to other cellular compartments, where NO-stimulated phosphodiesterase could be activated separately from 8-iso-PGF2╬▒-stimulated cAMP. Thus, 8-iso-PGF2╬▒ might interact with NO at the level of cAMP or cGMP.
The pathomechanism whereby 8-iso-PGF2╬▒ prevents NO-inhibited JCl might be observed under several conditions characterized by increased blood pressure, such as hypertension and chronic renal failure. In such conditions, urinary 8-iso-PGF2╬▒ 9 [29] and cAMP [30] are elevated as well as other oxidative stress mediators. On the contrary, NO production is impaired in part due to limitations on substrate (L-arginine) availability and increased circulating levels of endogenous NO synthase inhibitors, in particular asymmetric dimethylarginine and reduced renal cortex abundance of the neuronal NOS [31]. This effect of NO correlates with urinary cGMP level [32]. This pathological condition is characterized by sodium retention as a consequence of enhanced sympathetic activity [33] and increase synthesis of intrarenal angiotensin II synthesis [34], endothelin-1 [35], and 8-iso-PGF2╬▒, all of which stimulate sodium reabsorption. In contrast, the antinatriuretic effect of NO is blunted under these conditions, leading to sodium retention, hypertension, fibrosis, and end-organ damage.
The beneficial effect of 8-iso-PGF2╬▒ blockade has been observed in several animal models. Studies in diabetic animals with nephropathy showed that supplementation with vitamin E via reduced production of transforming growth factor-beta reduced urinary isoprostane level and improved proteinuria and blood urea nitrogen level [36]. In spontaneously animal models of hypertension and angiotensin II-induced hypertension, both with elevated plasma isoprostane level, Tempol administration decreases renal isoprostane excretion and lowers blood pressure [4,37].
These beneficial effects observed by isoprostane blockade (and increase in NO availability) can prevent sodium overload, hypertension, chronic fibrosis, and end-organ damage. Clinical evaluations are under review to estimate the beneficial effect of isoprostane blockade (NCT03358524, NCT01125501, and NCT00552227).
Although the results of this report can explain important pathophysiological conditions, it has some limitations. One important limitation is the H89, despite being the most commonly used PKA inhibitor, might have nonspecific inhibitory effects.
In conclusion, 8-iso-PGF2╬▒ can increase JCl at the kidney cTAL via stimulation of PKA even when JCl is inhibited by NO. This increase of JCl is associated with a higher cAMP tissue concentration, which cannot be inhibited by NO, indicating the importance of 8-iso-PGF2╬▒ as a therapeutic intervention in kidney repair and functional restoration to regulate fluid and electrolyte balance.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









