Pleotropic effects of hypoxia-inducible factor-prolyl hydroxylase domain inhibitors: are they clinically relevant?
Article information

Abstract
Anemia is common in patients with chronic kidney disease (CKD) and is mainly caused by insufficient production of erythropoietin from fibrotic kidney. Because anemia impairs quality of life and overall prognosis, recombinant human erythropoietin-related products (erythropoiesis-stimulating agents, ESAs) have been developed to increase hemoglobin level for decades. However, many safety concerns have been announced regarding the use of ESAs, including an increased occurrence of cardiovascular events, vascular access thrombosis, cancer progression, and recurrence. Hypoxia-inducible factor (HIF) is crucial to erythropoietin production, as a result, prolyl hydroxylase domain (PHD) enzyme inhibitors have been new therapeutic agents for the treatment of anemia in CKD. They can be administered orally, which is a preferred route for patients not undergoing hemodialysis. In clinical trials, PHD inhibitor could induce noninferior effect on erythropoiesis and improve functional iron deficiency compared with ESAs. Although no serious adverse events were reported, safety is still a concern because HIF stabilization induced by PHD inhibitor has pleotropic effects, such as angiogenesis, metabolic change, and cell survival, which might lead to unwanted deleterious effects, including fibrosis, inflammation, cardiovascular risk, and tumor growth. More molecular mechanisms of PHD inhibition and long-term clinical trials are needed to observe these pleotropic effects for the confirmation of safety and efficacy of PHD inhibitors.
Introduction
Anemia is a major complication of chronic kidney disease (CKD) and impacts patients overall outcomes and quality of life [1]. Impairment of synthesis of erythropoietin from the pericytes in fibrotic kidney primarily contributes to renal anemia [2]. Therefore, the use of recombinant human erythropoietin related products (erythropoiesis-stimulating agents, ESAs) has been the major therapeutic strategy for over three decades [3]. The prevalence rate of anemia rises up to 90.2% in CKD stage 5 [4]. In a CKD registry with 14,415 patients, anemia occurred in 60% of nondialysis and 93% of dialysis-dependent patients. More ESAs were administered in dialysis-dependent patients than in nondialysis patients (82% vs. 24%) [5]. However, ESA therapy is associated with cardiovascular adverse effects, including hypertension and thromboembolism in some studies, especially when using ESAs to target a hemoglobin level of greater than 11 g/dL [6–8]. Cancer progression or recurrence is also a significant concern with ESA therapy, and the 2012 Kidney Disease: Improving Global Outcomes (KDIGO) Clinical Practice Guidelines for anemia in CKD recommended using ESA therapy with caution in those with current or previous malignancy [9].
Anemia results in tissue hypoxia, and hypoxia-inducible factor (HIF) is a crucial transcription factor by activating the hypoxia-responsive genes, including erythropoietin. HIF is a heterodimer that consists of an α and β unit. This heterodimer translocates to the nucleus and binds to DNA sequences named hypoxia response elements. The β unit is present consistently (HIF-β), while there are three isoforms of α subunit (HIF-1α, HIF-2α, and HIF-3α) whose activity is primarily controlled by their degradation rate. In normoxia, HIF-α subunits are hydroxylated by a family of prolyl hydroxylase domain (PHD) enzymes, of which there are three isoforms: PHD1, PHD2, and PHD3. The hydroxylated HIF-α subunits are degraded by the von Hippel-Lindau tumor suppressor (VHL)-mediated ubiquitination [10]. Under hypoxia, the hydroxylation activity of PHDs is suppressed, which results in HIF-α accumulation and combines with HIF-β, leading to increased transcription of hypoxia-responsive genes [11]. Elucidation of these mechanisms led to the development of PHD inhibitors as promising therapeutic agents to stabilize HIF and correct anemia in CKD. Currently, several PHD inhibitors including roxadustat, vadadustat, daprodustat, molidustat, enarodustat, and desidustat have been developed and undergoing clinical trials.
Nevertheless, in addition to erythropoietin, more than 60 direct target genes of HIFs have been identified [12]. Therefore, in addition to erythropoiesis, HIF also plays key roles in angiogenesis, energy metabolism, immune modulation, cell proliferation, and survival (Fig. 1). As a result, pleotropic effects of HIF stabilization may be concerned during the treatment with PHD inhibitor. In this review, we will discuss these pleotropic effects and clinical relevance. Through investigating the molecular mechanisms and clinical evidence of pleotropic effects of PHD inhibition, we hope to avoid deleterious off-target effects and develop therapeutic agents with more safety and efficacy.
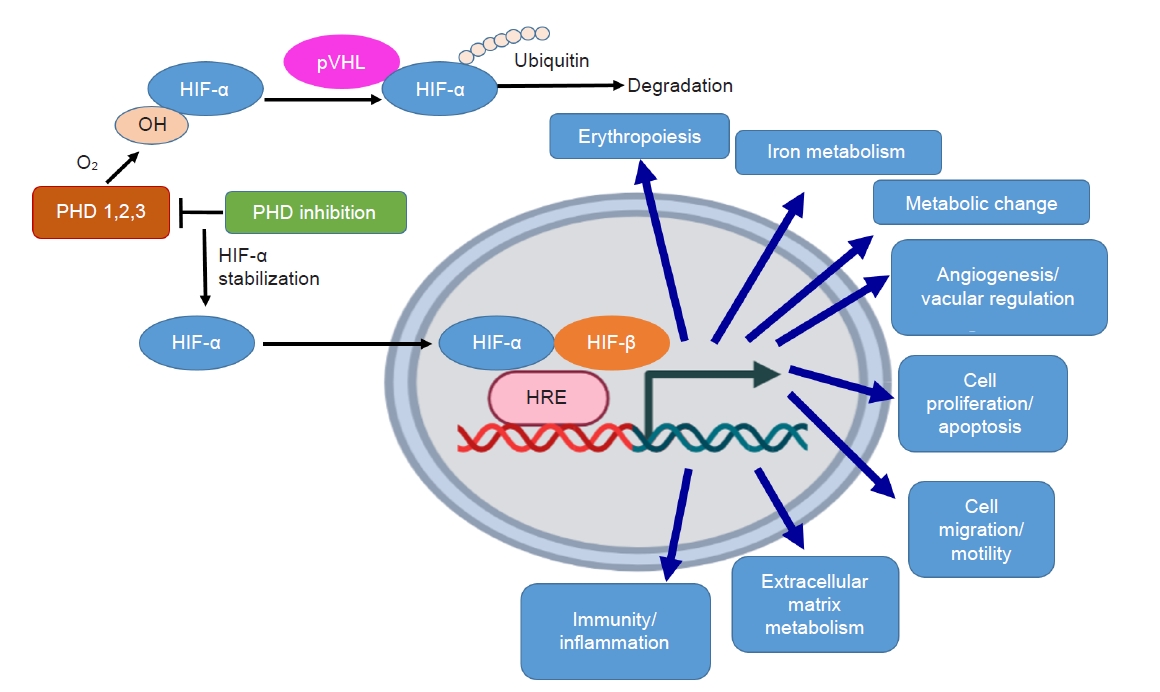
PHD inhibition stabilizes HIF-α and results in various pleotropic effects.
In normoxia, HIF-α is hydroxylated (OH) by PHD enzymes. OH HIF-α is ubiquitinated by the von Hippel-Lindau (pVHL) ubiquitin ligase, resulting in degradation by the proteasome. Under PHD inhibition, HIF-α hydroxylation is inhibited and is translocated to the nucleus where it dimerizes with HIF-β, binds to the hypoxia response element (HRE), and induces pleotropic effects.
HIF, hypoxia-inducible factor; PHD, prolyl hydroxylase domain.
The pleotropic effects of prolyl hydroxylase domain inhibition
Iron metabolism
PHD inhibitors can increase hemoglobin levels by decreasing hepcidin and ferritin levels and decrease transferrin saturation by increasing total iron-binding capacity. Through inhibition of ferroportin (FPN), hepcidin inhibits iron absorption from intestine and impedes the release of iron from liver and reticuloendothelial macrophage. The hepcidin levels are elevated in CKD patients due to decreased renal clearance and upregulated systemic inflammation, which results in the malabsorption and impaired availability of iron, contributing to renal anemia [13]. In an animal study, HIF stabilization in global Vhl knockout mice could suppress the hepcidin gene (Hamp1) messenger RNA levels in the liver via increasing erythropoietic activity and serum growth differentiation factor 15 level. Reduced iron and ferritin levels were noted as well [14]. Another murine model showed that increase of HIF-2α, not HIF-1α was essential for FPN activation in intestinal cells under conditions of low iron [15]. And experiments of intestinal cell line demonstrated that PHD enzymes were downstream of hepcidin-FPN pathway in the regulation of HIF-2α, and pharmacological inhibition of PHD could stabilize HIF-2α [16].
Clinical study of PHD inhibitor (roxadustat) in nondialysis CKD or dialysis-dependent patients, hepcidin decreased more significantly in roxadustat-treated group than in placebo or ESA group [17,18]. In study of daprodustat and vadadustat in dialysis-dependent patients, the hepcidin levels were also lower in PHD inhibitor group than in darbepoetin alfa group [19,20]. As a result, PHD inhibitors might decrease the use of iron supplement to avoid the risk of anaphylactic reaction and the adverse effects such as increase of oxidative stress or iron overload.
Metabolic change
In obese type 2 diabetic mice, the enarodustat-treated group had lower body weight compared with vehicle group without the difference in the amount of food intake between groups. Enarodustat treatment also improved insulin sensitivity and glucose tolerance [21]. A similar improvement was reported in another murine model of Hif-p4h-2–hypomorphic mice (partial loss of Phd2 function), whether fed a normal or high-fat diet. These mice had higher HIF levels because of PHD deficiency and higher levels of glucose transporter 2 (GLUT2) in the liver as well as glycolysis enzymes in the skeletal muscle and white adipose tissue (WAT), contributing to the improved glucose tolerance. In addition, decreased size of adipocytes, a reduced number of adipose tissue macrophages, and increased glucose transporter GLUT4 expression were noticed in the Hif-p4h-2–hypomorphic mice, which seemed likely the mechanisms of increased insulin sensitivity. After administration of another PHD inhibitor, FG-4497, to wild-type mice with metabolic dysfunction, these beneficial effects could be reproduced, indicating that this is a plausible class effect of PHD inhibitors [22]. HIF-1α is crucial for islet β cell function. Deletion of HIF-1α in β cells caused glucose intolerance in mice due to impaired first-phase insulin release [23]. The increase of HIF-1α expression during exercise in skeletal muscle was important for glucose metabolism and insulin action by maintaining the translocation of GLUT4 to the cell surface [24]. Additionally, HIF-2α stabilization increased Irs2 and cyclic AMP-specific phosphodiesterase gene expression, leading to upregulated and downregulated insulin and glucagon signaling, respectively. Human and mice hepatocyte-specific deletion of HIF-2α markedly attenuated these effects of PHD inhibitors, showing the effect of PHD inhibition on glucose metabolism was mainly HIF-2α dependent in the liver [25]. However, this beneficial effect on glucose control of diabetic patients has not yet been determined in clinical trials.
Regarding lipid metabolism, the levels of plasma total cholesterol and the mass of WAT were lower, while adiponectin levels were higher in the enarodustat group of obese type 2 diabetic mice [21]. Restoration of adiponectin by enarodustat was associated with the reduction of adipocyte hypertrophy, supporting the results of the protective roles of adipokine against metabolic syndrome [26]. Similarly, Hif-p4h-2–hypomorphic mice had reduced serum cholesterol and were protected against hepatic steatosis under a high-fat diet. Moreover, decreased conversion of pyruvate to acetyl coenzyme A (acetyl-CoA) as a result of an increased Pdk1 expression in skeletal muscle, WAT, and liver was bound to be a probable cause of the amelioration of de novo lipogenesis [22]. In a clinical trial, roxadustat lowered low-density lipoprotein (LDL) cholesterol levels in nondialysis CKD patients and this effect developed 4 weeks after treatment and was maintained for up to 1 year [27]. In dialysis patients, roxadustat lowered the cholesterol including total cholesterol, LDL, triglyceride, and LDL to high-density lipoprotein ratio [17]. In a 4-week treatment, daprodustat also decreased cholesterol with dose-dependent effect in dialysis and nondialysis patients [28].
Acute kidney injury
HIF is a master regulator of energy metabolism, inflammation, and cell proliferation, contributing to the protective role of acute kidney injury (AKI). Hill et al. [29] showed that either hif1α+/– and hif2α+/– mice had higher injury score and worse renal function in the ischemia-reperfusion injury (IRI) model. They also found that PHD inhibitor L-mimosine administered 6 hours before IRI could protect mouse kidneys from injury. HIF-1α was detected predominantly at the corticomedullary junction as well as distal tubular cells and was obviously increased, while HIF-2α increased expression mainly in interstitial cells at the corticomedullary junction compared with control group [29]. On the other hand, in a model of ischemia produced by oxygen-glucose deprivation of cultured renal proximal tubule cells, the cell viability was increased and the levels of reactive oxygen species (ROS) were reduced significantly by enarodustat treatment or small interfering RNA (siRNA) knockdown of PHD2, but not of PHD1 or PHD3. These protective effects were mediated by the increase of glycogen storage and the upregulation of genes of glycogen synthesis by HIF-1α before injury. Oxidative stress could be attenuated because sufficient glycogen produced reduced nicotinamide adenine dinucleotide phosphate that reduced glutathione, and ROS was then scavenged by reduced glutathione. In an IRI model of rat, pretreatment with the PHD inhibitors could upregulate glycogenesis, prolong glycolysis, and delay adenosine triphosphate consumption, contributing to renal protection [30]. For endothelial cell, specific inactivation of endothelial PHD2 attenuated IRI-AKI. Although PHD2 inhibition was not sufficient to induce detectable HIF activity in the kidney endothelium, the therapeutic effect was dependent on HIF-1α but not HIF-2α and was generated by suppressing the expression of proinflammatory genes and recruitment of neutrophils and macrophage [31]. Moreover, hyper-methylation of pericytes in the kidney was noted after AKI and was the major cause of ensuing CKD [32], whether PHD inhibitors influence methylation after AKI should also be investigated in the future. Taken together, PHD inhibitors have the potential to prevent AKI clinically. AKI frequency and severity could be monitored in long-term PHD inhibitor users. Clinical trials of the preventive effects by PHD inhibition for AKI could be performed in patients with operation (e.g., cardiac surgery associated-AKI).
Renal fibrosis and chronic kidney disease progression
PHD inhibition effect on renal fibrosis is still controversial and possibly cell-dependent. Early study demonstrated that mice in a 5/6 nephrectomy model with VHL deletion in tubular epithelial cells could stabilize HIF-1α but contribute to renal fibrosis. The data showed the increase of fibroblasts number and collagen production in VHL-deleted mice, and also had higher albuminuria and profibrotic gene expression, including transforming growth factor (TGF)-β1, plasminogen activator inhibitor-1, and connective tissue growth factor [33]. In cultured renal medullary interstitial cells, angiotensin II remarkably increased HIF-1α levels, which induced the accumulation in collagen I/III and tissue inhibitors of metalloproteinases (TIMP)-1 protein levels. HIF-1α siRNA rather than HIF-2α siRNA completely blocked the effects of angiotensin II on collagen I/III and TIMP-1 production. HIF-1α siRNA also attenuated angiotensin II-induced elevation of proliferating cell nuclear antigen and vimentin, a marker of cell proliferation and cell transdifferentiation, respectively. In addition, it was reported that overexpression of PHD2 gene attenuated angiotensin II-induced profibrotic action and silencing of PHD2 gene enhanced angiotensin II-induced profibrotic action [34].
Nevertheless, recent studies showed neutral results of PHD inhibition on renal fibrosis. The study deleted PHD1, 2, 3 in renal erythropoietin-producing cells and found no difference of collage production compared to control group [35]. There was also no significant increase of profibrotic gene and most inflammatory gene expression in mice with PHD deficiency compared to control group in the kidneys on day 7 and day 14 after unilateral ureteral obstruction surgery [35]. Furthermore, the concern for the impact of HIF in pericytes on kidney fibrosis arises from the fact that pericytes are the major precursor cells of myofibroblasts in fibrotic kidney. Previous study reported that Gli1 is one of the markers of pericyte and the ablation of Gli1+-cell attenuated renal fibrosis [36]. Hence, we induced Gli1+ cell-specific HIF stabilization via Vhl or Phd2 knockout showed increased serum erythropoietin and polycythemia without a significant difference in profibrotic gene expression and kidney fibrosis [37]. However, because there are many other markers of fibroblasts/pericytes such as platelet-derived growth factor (PDGF) receptor β, CD73, smooth muscle myosin protein, and tenascin-C which manifest as different lineages, we need further study to clarify the effect of HIF activation in pericytes from other lineages in various models (ischemia-reperfusion, adenine-induced CKD, aristolochic acid nephropathy, and diabetic nephropathy) and clinical trials to observe the effect of PHD inhibitors on renal fibrosis [38].
For CKD progression, a diabetic mice model showed that enarodustat-treated mice also exhibited reduced albuminuria and amelioration of glomerular epithelial and endothelial damage compared with the vehicle group. In vitro experiments demonstrated that reduced C-C motif chemokine ligand 2/monocyte chemoattractant protein-1 (CCL2/MCP-1) expression in mesangial cells was contributed by both local HIF-1α activation and restoration of adiponectin [21]. However, clinical reports of renal outcome in patients using PHD inhibitors were rare, only a 3-year follow-up study of patients with daprodustat treatment showed no impact on CKD progression [39].
Immunity and inflammation
For innate immunity, HIF-1α and HIF-2α have been shown to express both in macrophages and neutrophils. In hypoxia, HIF-1α is essential for neutrophil function and survival [40]. Thompson et al. [41] found that the neutrophils from the blood and lungs of mice or zebrafish with acute lung injury showed early expression of HIF-1α. In contrast, HIF-2α was expressed later and maintained during the resolution phase of acute lung injury. Through the study of patient cells and in vivo in a zebrafish model, HIF-2α gain-of-function mutations resulted in a reduction in neutrophil apoptosis and promoted the persistence of neutrophilic during chronic inflammation [41]. Moreover, Sadiku et al. [42] identified PHD2 as the dominant HIF-hydroxylase in neutrophils. Myeloid-specific genetic deletion of Phd2 or pharmacological inhibition of PHD2 activity led to robust neutrophilic inflammation in response to Streptococcus pneumonia and in a lipopolysaccharide (LPS)-induced acute lung injury model. This exaggerated inflammatory response was due to increased glycolytic flux and glycogen stores in neutrophil, which enhanced neutrophil functional capacity, motility, and survival [42]. Unlike PHD2, loss of PHD3 in neutrophils was associated with reduced inflammation because of the unique role for PHD3 in prolonging neutrophil survival during hypoxia [43].
Regarding macrophage, HIF-α isoforms crucially regulate macrophage function via metabolic change. HIF-1α was induced by M1 signals such as interferon gamma (IFN-γ) and LPS, whereas M2 signals such as interleukin (IL)-4 and IL-13 favored HIF-2α expression. HIF-1α mediated a metabolic switch from oxidative phosphorylation (OxPhos) to glycolysis and was crucial for macrophage phagocytosis and inducible nitric oxide synthase (NOS) generation. Conversely, HIF-2α enhanced the M2 gene arginase 1 expression and polarized an M2 macrophage phenotype [44]. In contrast to the findings in neutrophilic inflammation, loss of PHD3 resulted in increased mortality in a murine model of LPS-induced peritonitis, where inflammation was predominantly induced by macrophage [45]. Enarodustat reduced expression of CCL2/MCP-1 and macrophage infiltration in diabetic mice [21].
For adaptive immunity, HIF-1α activation favored T helper 17 cells (Th17) development over regulatory T cell (Treg) differentiation via enhancing glycolytic activity, upregulation of the transcription factor retinoic acid-related orphan receptor gamma t, and degradation of the Treg transcription factor FOXP3 [46,47]. And mammalian target of rapamycin (mTOR) signaling was essential to induce HIF-1α expression in Th17 cells [46]. On the other hand, a recent study investigating the role of microRNAs in Tregs also identified that Treg cell development could be suppressed by both HIF-1α and HIF-2α, demonstrating similar roles of the HIF protein in Treg differentiation [48]. Furthermore, PHD3 was found to be upregulated in Tregs and required for the development of Tregs. The PHD3 inhibitors dimethyl oxalylglycine or siRNAs, which upregulated HIF-1α, could down-regulate Foxp3 expression, and enhanced the development of Th17 cells [49]. Similar to CD4+ T cell, CD8+ T cell differentiation and activation required upregulation of HIF-1α and metabolic adaptations, including a switch from OxPhos to glycolysis. HIF-1α enhanced viral and tumor cell clearance, but was also associated with excessive inflammation and mortality in chronic viral infections [50,51]. Finlay et al. [52] also showed that mTOR complex 1 (mTORC1)-mediated expression of HIF-1α was required not only to sustain glycolysis in CD8+ T cells but also to generate inflammatory mediators such as cytolytic effector molecules, including perforin, granzymes, and IFN-γ.
Regarding B cell population, it was found that HIF-1α was stabilized following B cell receptor activation, and that B cell-specific deletion of Hif resulted in fewer IL-10–producing B cell population and exacerbated collagen-induced arthritis and experimental autoimmune encephalomyelitis. And HIF-1α–dependent glycolysis facilitated CD1dhighCD5+ B cells expansion [53]. However, sustained hypoxia or HIF-1α stabilization by B cell-specific Vhl deletion inhibited mTORC1 activity in germinal center B cell, reduced antigen-specific germinal center B cells, undermined class-switching, and reduced generation of early memory B cells [54].
Overall, these data on immune cells highlight that, even within a single cell population, targeting HIFs or PHDs can have highly complex and variable outcomes of infection, inflammation, and resolution. The final outcome of HIF activation in an inflammatory response depends on various factors, including which cell and tissue type are affected, which HIF or PHD isoform is targeted, and other environmental factors such as the cytokine profile, metabolite availability, tissue-specific factors, and duration and severity of hypoxia [55].
In clinical trials, upper respiratory infection, pneumonia, and urinary tract infection were more common in roxadustat than in control group [18,56], which might be due to immune modulation by PHD inhibition. Herein, we should continue monitoring the incidence of infection rate under the use of PHD inhibitors, and discuss the discontinuation of inhibitors if patients are undergoing any infection.
Cancer development and progression
Because most solid cancers are in hypoxia environment and HIF expression is high, cancer development and progression are the major concern in PHD inhibitor users. HIF-1α acts as a versatile regulator that is involved in crucial aspects of cancer biology, including angiogenesis, glucose metabolism, cell survival, resistance to cell death, and tumor metastasis. In most cases, HIF-1α overexpression is associated with treatment failure and increased mortality, and inhibitors of HIF-1α have been studied as anticancer therapeutics [12].
HIF-1α induced metabolic reprogramming in tumors with shifting OxPhos to aerobic glycolysis (also called the Warburg effect) for their energy production [57,58]. Additionally, HIF-1α could inhibit acetyl-CoA dehydrogenases and fatty acid β-oxidation, which was beneficial for cancer proliferation via enhanced Glut2 expression and glycolysis, reduced ROS production, and activation of prosurvival cancer signaling pathway [59]. These metabolic reprogramming effects provide sufficient energy for tumor growth and metastasis even in hypoxia microenvironment. Of note, HIF-1α also upregulates proangiogenic genes encoding NOS, vascular endothelial cell growth factor (VEGF), and PDGF-B to augment vascular density and decreases O2 diffusion distances [60]. In addition, HIF-1α facilitated cancer cell proliferation by activating numerous growth factors and pathways such as insulin-like growth factor-2 [61], TGF-α [62], phosphoinositide 3-kinases, and mitogen-activated protein kinase pathways [63]. HIF-1α deterred apoptosis by activating the expression of the inhibitor of apoptosis protein 2 and inducing G1/S arrest through the upregulation of protein phosphatase 2A [64,65]. Nevertheless, HIF-1α also mediated hypoxia-related apoptosis by increasing the expression of Bcl-2 binding proteins 3 thereby inhibiting the antiapoptotic effect of Bcl-2 [66], or by stabilizing wild-type p53 [67,68]. The function of HIF-1α in the regulation of apoptosis by hypoxia is complex and should be further investigated. Furthermore, a recent study showed that long noncoding RNA (lncRNA) DANCR with the NF90/NF45 complex could stabilize HIF-1α, leading to increased cancer proliferation and metastasis in nasopharyngeal carcinoma [69]. Another lncRNA, MTA2 transcriptional regulator RNA, functioned as a promoter and enhanced the proliferation and metastasis of pancreatic cancer cells by enhancing the stability of HIF-1α protein via metastasis-associated protein 2-induced deacetylation [70]. On the other hand, clear cell renal cell carcinoma (ccRCC) has been reported to develop in approximately 70% of patients with VHL disease [71]. HIF-2α has been implicated tumorigenesis in VHL-reconstituted ccRCC [72], and HIF-2α inhibitor has been tested in phase 2 clinical trials for ccRCC in VHL disease [73].
Current PHD inhibitors have different impact on PHD isoforms, contributing to variable effect on HIF stabilization. These inhibitors have been tested in various cancer models, resulting in positive and negative outcomes. For example, roxadustat inhibited all PHD isoforms and reduced tumor growth of macrophage-abundant tumors via normalization of tumor vessels [74,75]. Sensitivity to chemotherapy was repaired by roxadustat as well in a murine tumor model [76]. Vadadustat had a major impact on PHD3 and PHD2, resulting in higher upregulation of HIF-2α than HIF-1α without increase of plasma VEGF in patients [77]. Daprodustat influenced PHD1 more than other isoforms. High dose of daprodustat did not pose carcinogenic risk in vivo [78]. The results from the ASCEND-ND trial demonstrated that daprodustat for the treatment of anemia in patients with CKD not receiving dialysis had higher risk of cancer-related death or tumor progression or recurrence compared to the group of darbepoetin alfa [39]. However, another post-hoc modified intention-to-treat analysis of pooled data from the ASCEND-D and ASCEND-ND trials showed that daprodustat was not associated with an increased risk of cancer, or cancer mortality, relative to ESA [79]. Molidustat inhibited PHD2 more than other isoforms and inhibited breast cancer cell survival, self-renewal capacity, and enhanced the efficacy of chemotherapeutic gemcitabine [80]. Overall, HIF-1α stabilization is required for tumor growth and metastasis. Nevertheless, aforementioned findings indicate that under certain circumstances, HIF-1α stabilization may exert protective effects and even decrease tumor cell aggressiveness. Different cancer showed distinct PHD isoform expression, and the impact of the PHDs on tumor progression is diverse and cell-dependent. These studies highlight that longer follow-up data period and more patients with dialysis or nondialysis are desirable to evaluate the impact of PHD inhibitors on cancer development and progression compared to that of ESAs. Furthermore, head-to-head comparison of different PHD inhibitors should be performed in the future not only for the investigation of efficiency of erythropoiesis but also for the impact on cancers.
Cardiovascular risk and atherosclerosis
According to previous studies, HIF-1α stabilization could reduce lipogenesis [22], which might ameliorate atherosclerosis. However, the pathogenesis of atherosclerosis includes the dysfunction of inflammatory signaling pathway as well [81]. Some studies displayed that HIF-1α activation contributed to atherosclerosis. One finding was that the upregulation of nuclear factor kappa B, VEGF, and ROS by HIF-1α led to endothelial cell dysfunction contributing to the progression of atherosclerosis [82]. Another mechanism was that HIF-1α regulated macrophages and promoted inflammation and migration of macrophages. HIF-1α also promoted the formation of foam cells by reducing the ABCA1 cholesterol efflux protein [82]. This along with smooth muscle migration and proliferation contributed to plaque growth and atherosclerosis [83]. Furthermore, pulmonary arterial hypertension (PAH) may contribute to cardiovascular risk as well. It had been found that endothelial HIF-2α was required for the increase of the expression of vasoconstrictor molecule endothelin-1 and a concomitant decrease in vasodilatory apelin receptor signaling, contributing to the development of increased pulmonary artery pressures [84]. PHD2 plays a crucial role in the mechanisms of severe PAH. PHD2-deficient endothelial cells were demonstrated to promote smooth muscle cell proliferation in part through HIF-2α-induced C-X-C motif chemokine 12 expression [85].
Regarding cardiovascular risk in clinical trials, roxadustat demonstrated a similar cardiovascular safety profile and overall mortality compared with ESAs in a pooled analysis of cardiovascular safety of dialysis-dependent CKD [86]. In subgroup analysis, patients who received roxadustat compared with ESA in the incident dialysis subgroup had a lower risk of all-cause mortality as well as time to the first cardiovascular event than those in the stable dialysis subgroup [86]. In another roxadustat study in nondialysis CKD patients, there was no difference in cardiovascular risk between roxadustat and placebo group [56]. A recent study of daprodustat in nondialysis patients also showed no difference in the major adverse cardiovascular event (MACE), thromboembolic event, hospitalization for heart failure, death from any cause between daprodustat and darbepoetin alfa group [39]. Moreover, a meta-analysis from nine randomized controlled trials in a total of 8,022 enrolled patients revealed that there was no increase in cardiovascular risk of daprodustat treatment in patients undergoing dialysis or not [87]. There was also no difference in MACEs between molidustat and darbepoetin treatment groups [88]. More importantly, although there was no increase in cardiovascular risk of vadadustat treatment in patients with incident or maintenance dialysis of the INNO2VATE trial [89], vadadustat treatment in patients with non–dialysis-dependent CKD of the PRO2TECT trial resulted in significant higher hazard ratio of MACE, which failed to prove cardiovascular safety of vadadustat [90]. In addition to cardiovascular adverse effect, thromboembolism is also a concern of PHD inhibitors. Pilli et al. [91] showed that stabilization of HIF-1α in the liver was associated with reduced protein S (PS) expression and a lower plasma PS level, suggesting increased likelihood of thrombosis. Animal model study showed that hypoxia contributed to intimal hyperplasia formation in arterial prosthetic grafts [92,93]. Another animal model study also demonstrated that HIF-1α expression was associated with the degree of intimal hyperplasia in the venous grafts possibly by inducing the switch of adventitial fibroblasts to myofibroblasts and migration into the neointima [94]. In PYRENEES study of roxadustat in dialysis patients, the percentage of arteriovenous fistula thrombosis event in roxadustat group was higher than ESA group (12.1% vs. 7.4%) [95]. Hence, the U.S. Food and Drug Administration expressed the concern about these potential adverse effects of PHD inhibitors in 2021 [96].
Summary and perspective
The studies described above highlight various pleotropic effects of HIF stabilization by PHD inhibitors. Different PHD inhibitors might have variable impact on each PHD isoform (PHD1, 2, and 3), resulting in different levels of HIF-1α or HIF-2α and subsequent pleotropic effects. Some pleotropic effects are beneficial but other unwanted effects should be avoided. Current evidence is summarized in Table 1. More efforts should be performed to elucidate what are the alternative hydroxylation targets of PHD inhibitors and whether isoform-specificity of PHD inhibitors can be improved for specific targeting rather than pan-suppression of PHD activity. The timing and dosing of PHD inhibition should also be tested to improve the specificity and side-effect profiles.

Current evidence of pleotropic effects by PHD inhibition
Clinically, we should evaluate the trade-off between erythropoietic effects and avoidance of adverse effects. Therefore, larger number of patients and longer observation period are required to evaluate pleotropic effects. In nondialysis patients, the effect of PHD inhibitors on CKD progression could be evaluated. If no significant MACEs are noticed in the future study, higher target hemoglobin level could be tested (near normal, >12 g/dL) for potential better quality of life and outcomes.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Funding
YHC was supported by Ministry of Science and Technology, Taiwan (MOST; 108-2314-B-002-060, 109-2314-B-002-188, and 110-2314-B-002-115), National Taiwan University Hospital (NTUH; 107-003974, 109-004470, and 110-P03), and the Mrs. Hsiu-Chin Lee Kidney Research Foundation. SYP was supported by MOST (106-2314-B-418-006, 107-2314-B-418-001, 108-2314-B-418-004, and 110-2314-B-418-002). SLL was supported by MOST (109-2314-B-002-260 and 110-2314-B-002-208), NTUH (111-S0014 and 111-TMU022), NTUH and National Taiwan University (NTU) College of Medicine (NSCCMOH-131-43 and 145-72), NTU (NTU-CC-110L893304 and 111L892704) and Taiwan Health Foundation.
Data sharing statement
The data presented in this study are available on request from the corresponding author.
Authors’ contributions
Conceptualization: YHC, SLL
Investigation: YHC
Supervision: SLL
Validation: SYP
Writing–original draft: YHC
Writing–review & editing: all authors
All authors read and approved the final manuscript.