



Introduction
Glomerular injury promoted by angiotensin II (Ang II) may arise from systemic and local vasoconstrictive hemodynamic or non-hemodynamic actions or from such effects of Ang II acting in concert [1ŌĆō3]. Hemodynamic actions include increased systemic arterial pressure and higher glomerular capillary pressure. Non-hemodynamic actions of Ang II include higher expression of assorted growth factors and proto-oncogenes, aberrant growth responses, inflammation exacerbation, fibrogenesis, pro-oxidative cytokines, and apoptosis [1ŌĆō5]. A non-hemodynamic action of Ang II, described in vascular smooth muscle cells and endothelial cells, is induction of oxidative stress that may arise from activation of NADH/NADPH oxidase (Nox) [6ŌĆō8], an enzyme that is also present in renal cells such as mesangial cells [9], podocytes [10,11], and proximal tubular epithelial cells [12]. Nox4 is the most abundant isoform of the Nox proteins in podocytes and promotes podocyte injury in diabetic nephropathy [13]. It has been well documented that excessive reactive oxygen species (ROS) can cause protein oxidation, lipid peroxidation, and DNA damage, leading to cell damage and apoptosis [14]. Ang II mediates apoptosis as well as oxidative stress via Ang II type 1 receptor (AT1R) in the kidney [2,3].
Podocytes play a critical role in maintaining the permselective function of glomerular filtration units [15,16]. The release of ROS causes proteinuria by affecting glomerular endothelial cells, podocytes, and basement membrane and by disturbing normal glomerular permeability [17,18]. The podocyte is a terminally differentiated cell, and severe injury to the cell causes apoptosis without regeneration [4,5].
In this study, using an in vitro model of cultured podocytes, we questioned whether Ang II induces oxidative stress in podocytes and investigated the underlying molecular mechanisms to determine whether such oxidative stress may induce podocyte apoptosis.
Methods
Mouse podocyte culture
Conditionally immortalized mouse podocytes were kindly provided by Peter Mundel (Goldfinch Bio) and were cultured and differentiated as described previously [19]. Briefly, to stimulate podocyte proliferation, cells were grown at 33 Ōäā (growth permissive conditions) in RPMI 1640 medium in the presence of 10 U/mL mouse recombinant ╬│-interferon (Roche). Subsequent experiments were performed with differentiated cells. For podocytes to acquire a differentiated phenotype, they were maintained at 37 Ōäā in 95% air/5% CO2 without ╬│-interferon (non-permissive conditions) for at least 2 weeks. All experiments were performed at least three times independently.
Treatment conditions and preparation of antibodies
Mouse podocytes were treated with Ang II (Sigma Chemical Co.) at the indicated concentrations and time durations in 50 ╬╝M of probucol (Sigma Chemical Co.) in 70% ethanol as an antioxidant. Primary antibodies used were goat anti-Nox4 (N-15) antibody (sc-21860; Santa Cruz Biotechnology Inc.), goat anti-╬▓-tubulin antibody (sc-9104; Santa Cruz Biotechnology), and polyclonal rabbit anti-rat zonula occludens (ZO)-1 (61-7300; Invitrogen).
Small interfering RNA for Nox4 and AT1R transfection
The podocytes were transfected by Nox4, AT1R small interfering RNAs (siRNAs), or negative control scrambled siRNA for 24 hours and incubated in the conditions stated above. One day before transfection, the culture medium was removed, and differentiated podocytes were cultivated in antibiotic-free RPMI 1640 medium supplemented with 10% FBS. Transfection began when cell confluence was 70% to 80%. Transient knockdown of Nox4 or AT1R was performed in mouse podocytes using Lipofectamine 2000 Transfection Reagent (Invitrogen) as per the introductions provided by the manufacturer. Briefly, Nox4 siRNA (sc-41587; Santa Cruz Biotechnology), AT1R siRNA (sc-29751; Santa Cruz Biotechnology), or control scrambled siRNA (Santa Cruz Biotechnology) were diluted into each six-well plate with transfection medium (Opti-MEM; Invitrogen) and incubated for 5 minutes. In parallel, lipofectamine diluted with Opti-MEM and siRNA were mixed, incubated at room temperature for 20 minutes, and then added to the cultured podocytes. After replacing the transfection mixture with RPMI 1640 medium after 5 hours, the inhibitory effect of siRNA was confirmed using western blotting analysis.
Indices of oxidative stress
Production of superoxide anion (O2ŌĆō┬Ę) was detected using a modification of the technique based on specific reduction of cytochrome C by superoxide anion in the media [20]. Briefly, after podocytes were exposed to RPMI 1640 without phenol red (GIBCO/BRL) with Ang II or probucol, cytochrome C (0.2 mg/mL, type II; Sigma Chemical Co.) was added to the media, and aliquot volumes of 300 ╬╝L were assayed at 2, 6, 12, or 24 hours of exposure time. The specific reduction of cytochrome C induced by superoxide anion was measured using a spectrophotometer at a wavelength of 550 nm at 25 ┬░C. Results are expressed as % relative to the control (condition without Ang II).
To assay superoxide dismutase (SOD) activity, the podocytes exposed to Ang II or probucol for 6, 12, and 24 hours were washed three times with ice-cold phosphate-buffered saline (PBS) and harvested in SOD assay buffer containing 2-amino-2-methyl-1,3-propanediol, boric acid, and diethylene triamine pentaacetic acid. The cells were disrupted by several freeze-thaw cycles, and the homogenates were clarified via centrifugation at 8,500├Śg for 10 minutes at 4 ┬░C. Clarified cell culture supernatants can generally be assayed without extraction. Protein content was measured in an aliquot of the homogenate using the method described by Lowry et al. [21]. The SOD activity was measured using BIOXYTECH SOD-525 (OxisResearch) and was evaluated in various conditions; the values were compared with those of the control.
Measurement of reactive oxygen species production
Intracellular ROS generation was assayed using the cell-permeable fluoroprobe 5- (and 6-)chloromethyl-2ŌĆÖ,7ŌĆÖ-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA; Molecular Probes). To examine the effect of Ang II on ROS generation, mouse podocytes were stimulated with 10ŌĆō6-M Ang II for 2 or 24 hours. To determine the effect of probucol or siRNA on Ang II-stimulated ROS generation, the mouse podocytes transfected with or without siRNA were co-treated with 100ŌĆō6-M Ang II or 50-╬╝M probucol. After treatment, cells were loaded with 10-mM CM-H2DCFDA, examined at 30 minutes using immunofluorescence microscopy, and immediately analyzed with an enzyme-linked immunosorbent assay reader at 480-nm excitation and 530-nm emission wavelengths. The fold changes were expressed as % of the control (condition without Ang II).
Mitochondrial ROS production was measured with MitoSOX Molecular Probes (M36008; Invitrogen), a red fluorescent dye that localizes to mitochondria. After treatment, live cells were washed three times with warm PBS and then incubated with 0.5-╬╝M MitoTracker Green (M7514; Invitrogen) and 5-╬╝M MitoSOX for 30 minutes at 37 ┬░C [22]. Cell fluorescence was observed using inverted immunofluorescence microscopy (FluoView FV10i; Olympus) at wavelengths of 488 nm for excitation and 594 nm for emission. A MitoSOX index was calculated as MitoSOX intensity per MitoTracker Green-positive area using ImageJ image analysis software (U.S. National Institutes of Health).
For detection of 8-oxo-2ŌĆ▓-deoxyguanosine (8-oxo-dG), a major form of oxidatively generated DNA damage, treated cells were fixed in 50% ethanol and then incubated with anti-8-oxo-dG antibody (4354-MC-050; R&D Systems). An 8-oxo-dG index was calculated as immunofluorescence intensity of 8-oxo-dG per individual cell using ImageJ image analysis software [22].
Immunofluorescence staining
Podocytes were cultured on cover slides and treated as indicated. The cells were washed twice with cold PBS and fixed for 10 minutes in 4% paraformaldehyde solution, followed by a 10-minute permeabilization in 0.2% Triton X-100 at room temperature. Thereafter, 10% goat serum was applied to block nonspecific binding for 1 hour at room temperature. Cells were then incubated with primary antibodies with a manufacturer-recommended dilution overnight at 4 ┬░C in the dark, followed by a 1-hour incubation with 1:200 (v/v) Alexa Fluor 488 (A32723; Invitrogen) for green and Alexa Fluor 594 (A32758; Invitrogen) for red conjugated secondary antibodies at room temperature. The nuclei were double stained with 4ŌĆÖ,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma Chemical Co.), and the images were viewed using a fluorescence microscope (TCS SP2 AOBS; Leica). Densitometry analyses of the fluorescence were conducted using Image J.
Western blotting analysis
The expression of Nox4 protein was assayed with western blotting analysis. The primary antibodies for anti-Nox4 antibody were diluted for western blotting analysis at 1:1,000. Total proteins were extracted from the podocytes using a protein extraction solution (PRO-PREP; Intron) containing leupeptin as a lysosomal inhibitor as per the manufacturerŌĆÖs protocols. The extracted protein concentrations were determined using a Bio-Rad kit (Bio-Rad Laboratories); 30 ╬╝g of boiled extract was loaded per lane on 10% SDS-PAGE gels and subsequently transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories). Thereafter, the membranes were air-dried, blocked in 5% fat-free dried milk, and incubated with the indicated primary antibodies overnight at 4 Ōäā. After three washes with Tris-buffered saline with 0.1% Tween, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase (Santa Cruz Biotechnology) for 1 hour at room temperature, and the resultant bands were visualized using the enhanced chemiluminescence system (Amersham Biotech Ltd.). To confirm equal loading, blots were reprobed with an anti-╬▓-actin antibody. In each experiment, the ratios of absorbance of each molecule vs. that of ╬▓-tubulin were analyzed and expressed as % of control (condition without Ang II).
Real-time polymerase chain reaction analysis
Total cellular RNA was extracted using TRIzol reagent (Invitrogen) from the treated podocytes. A total of 2 ╬╝g of RNA was reverse-transcribed into complementary DNA (cDNA) with the IScript cDNA Synthesis Kit (Bio-Rad Laboratories) as per the manufacturerŌĆÖs protocol. Real-time quantitative polymerase chain reaction (qPCR) was performed to evaluate the Nox4 messenger RNA (mRNA) level at the following conditions: 95 ┬░C for 30 seconds for annealing and 40 cycles of amplification (95 ┬░C for 15 seconds, 60 ┬░C for 30 seconds). The PCR reaction contained 1├Ś SYBR Green PCR Master Mix (Bio-Rad Laboratories), 1.5 ╬╝L of cDNA, and 0.2 ╬╝M of Nox4 primers (forward: 5ŌĆÖ-CCAGAATGAGGATCCCAGAA-3ŌĆÖ, reverse: 5ŌĆÖ-ACCACCTGAAACATGCAACA-3ŌĆÖ, NM_0053069). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal and loading control (forward: 5ŌĆÖ-CGAAGTCAACGGATTTGGTC-3, reverse: 5ŌĆÖ-AGCCTTCTCCATGGTGGTGA-3ŌĆÖ, NM_008084). The relative mRNA level of Nox4 vs. GAPDH was calculated using the 2-╬ö╬öCt method, and the fold-change was compared. All real-time qPCR reactions were performed in triplicate with three no-template negative controls for each primer pair, and the values were compared with those of controls.
Fluorescence-activated cell sorting assay
Podocyte apoptosis was determined using a fluorescein isothiocyanate (FITC)-conjugated annexin V/propidium iodide (PI) staining kit (BD Biosciences) following the manufacturerŌĆÖs instructions. In brief, treated podocytes were washed twice, collected (1 ├Ś 105/mL), and suspended with 100 ╬╝L of 1├Ś binding buffer (10-mM HEPES/NaOH pH 7.4, 140-mM NaCl, 2.5*mM CaCl2). Then, 5 ╬╝L of annexin V-FITC and 5 ╬╝L of PI were added for 15 minutes at room temperature in the dark. Thereafter, 400 ╬╝L of 1├Ś binding buffer was added, and the percentages of apoptosis and necrosis were determined using flow cytometry (FACSCalibur; BD Biosciences). All fluorescence-activated cell sorting (FACS) analyses for apoptosis detection after annexin V-FITC and PI labeling were performed three times independently under each experimental condition. The values were compared with those of controls.
Statistical analysis
The data represent at least three independent experiments. In ROS experiments, more than 30 podocytes in a given field in each independent experiment were examined for each condition. Data are representative images or expressed as mean ┬▒ standard deviation. Statistical significance was assessed using nonparametric Kruskal-Wallis analysis of variance analysis or Student t test using the IBM SPSS version 9.0 (IBM Corp.). The p-values of <0.05 were considered statistically significant.
Results
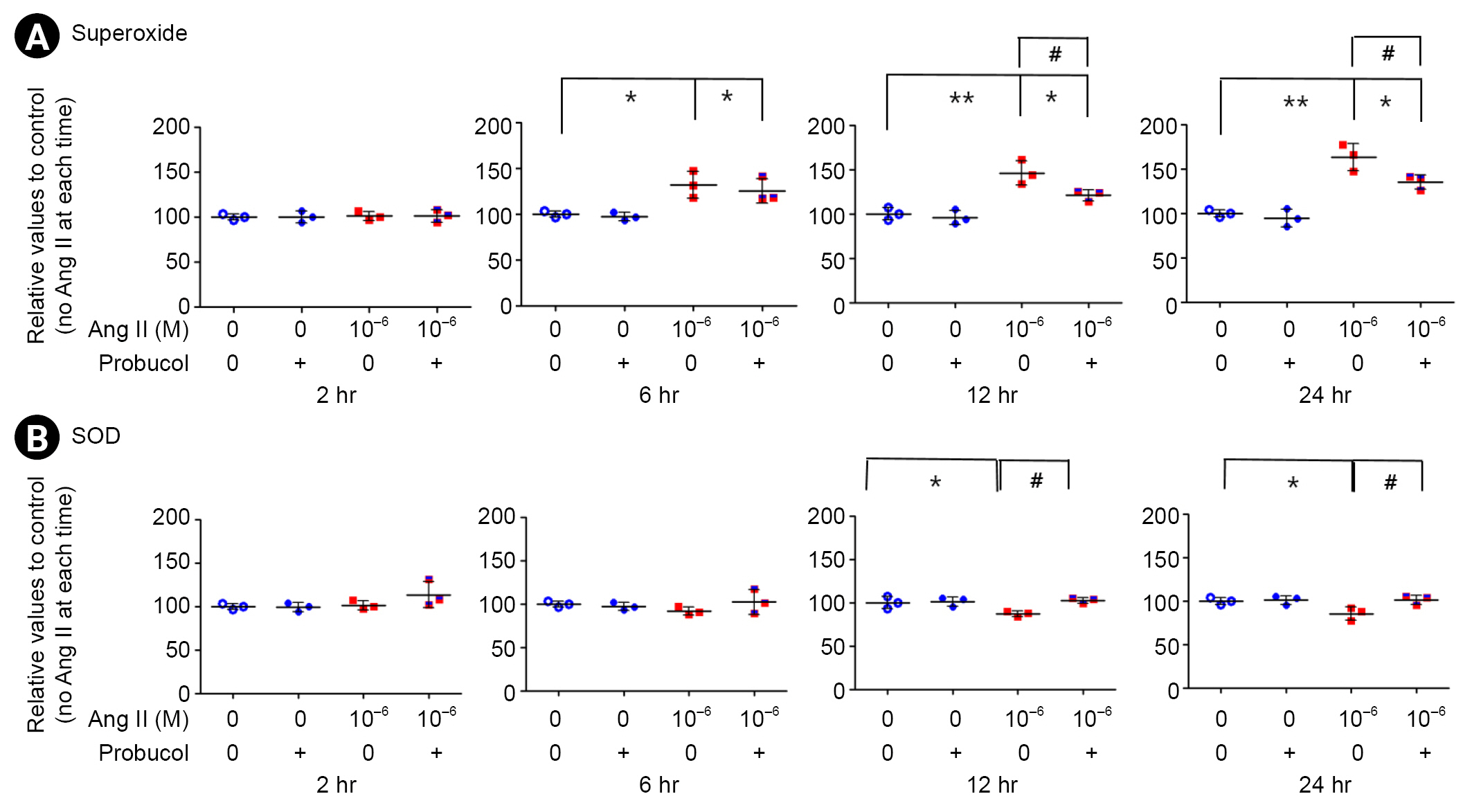
Angiotensin II-induced oxidative stress
Podocytes were incubated for 2, 6, 12, or 24 hours in media containing 10ŌĆō6-M Ang II or probucol. Administration of Ang II significantly increased the generation of superoxide anions within 6 hours, and there was a further increase of 63.3% at 24 hours (n = 3) (Fig. 1A; *p < 0.05 and **p < 0.01). However, probucol significantly suppressed Ang II-induced superoxide production (n = 3) (Fig. 1A; #p < 0.05). Administration of Ang II significantly suppressed SOD activity after 12 hours, and that was significantly recovered with probucol (n = 3) (Fig. 1B; *p < 0.05 and #p < 0.05).
Furthermore, we assessed the influence of Ang II on intracellular ROS levels using the CM-H2DCFDA. The podocytes were treated with various concentrations of Ang II (10ŌĆō8, 10ŌĆō7, and 10ŌĆō6 M) and incubated for 2 or 24 hours. Ang II increased ROS levels in dose- and time-dependent manners (n = 3) (Fig. 2A; *p < 0.05, #p < 0.05, and ##p < 0.01). However, probucol significantly suppressed the intracellular ROS levels at 24 hours (n = 3) (Fig. 2B; **p < 0.01 and ##p < 0.01).
Based on the ROS levels affected by Ang II in this study and our previous results of endoplasmic reticulum stress (ERS) levels by Ang II [23], we applied 10ŌĆō6 M of Ang II for subsequent analyses.
Next, we assessed the influence of Ang II on mitochondrial superoxide production using MitoSOX Red with MitoTracker Green and the accumulation of 8-oxo-dG in mitochondrial DNA. Untreated podocytes exhibited very low levels of MitoSOX Red fluorescence when cultured with or without antioxidants, whereas MitoTracker Green fluorescence was detected in cytoplasm (Fig. 2C, D). We quantified the MitoSOX index as MitoSOX intensity per MitoTracker Green-positive area using ImageJ. The quantitative data of MitoSOX index demonstrated that mitochondrial superoxide production was significantly higher in the Ang II-treated condition than in untreated conditions with or without probucol at 24 hours. When Ang II increased mitochondrial superoxide production by more than 2-fold, it was significantly recovered with probucol (n = 6) (Fig. 2C, D; *p < 0.05, **p < 0.01, and ##p < 0.01). We then examined 8-oxo-dG immunoreactivity and found that cytoplasmic 8-oxo-dG immunoreactivity was significantly increased in the Ang II-treated condition by 2-fold compared with that in the untreated conditions with or without probucol, and that was significantly recovered with probucol (n = 6) (Fig. 2E, F; *p < 0.05, **p < 0.01, and #p < 0.05). Taken together, these data suggest that Ang II induces mitochondrial oxidative stress and oxidative DNA damage in podocytes via downregulation of SOD activity, which is recovered with the antioxidant probucol.
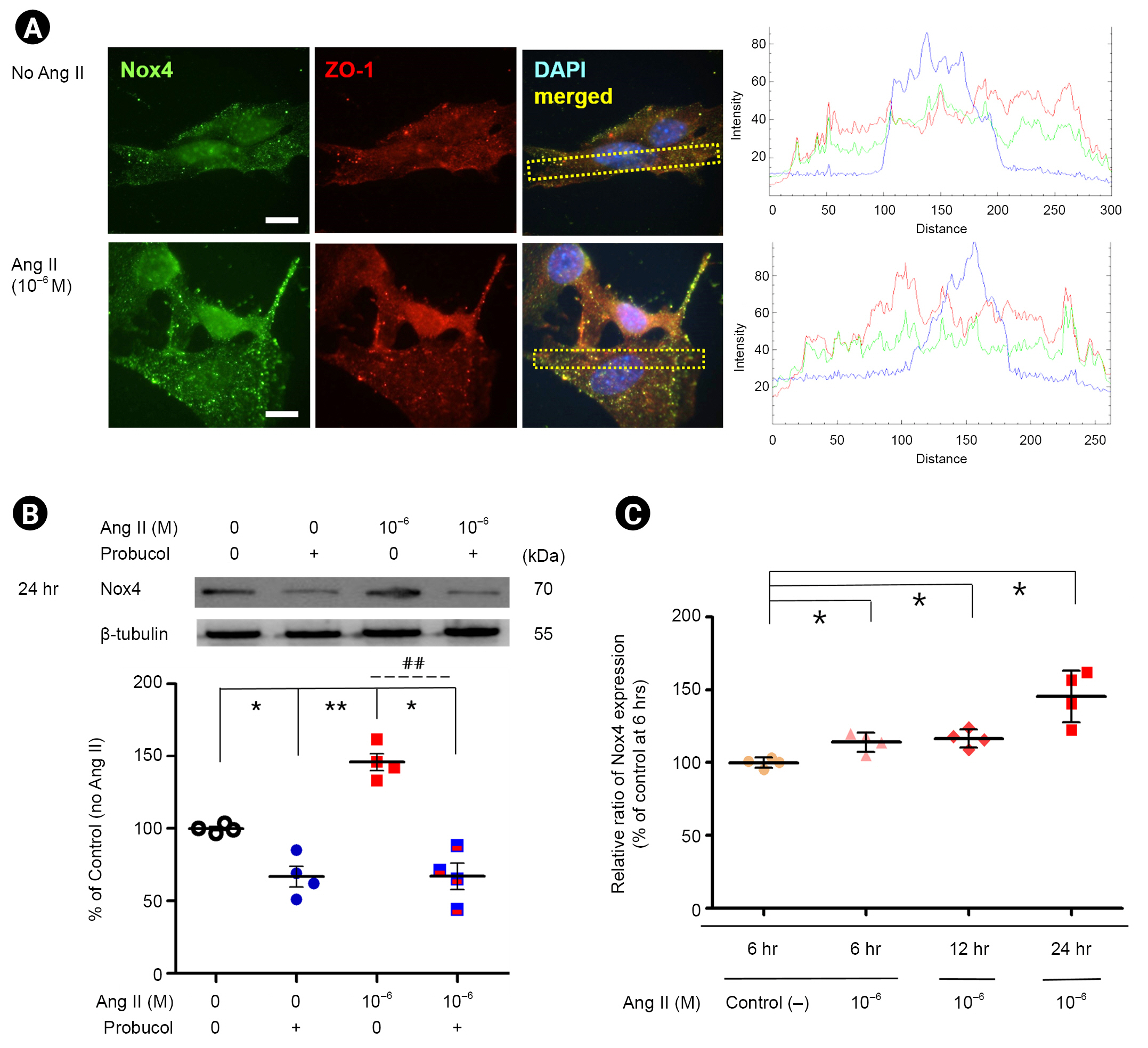
Angiotensin II increases Nox4 expression in podocytes
The distributions of Nox4 and ZO-1 were assessed with indirect immunofluorescence staining in Ang II-treated podocytes at 24 hours of incubation. Nox4 and ZO-1 were co-localized, and 10ŌĆō6M of Ang II appeared to concentrate both molecules and increase the intensity of Nox4 (Fig. 3A), as confirmed with western blotting and real-time PCR analysis.
To determine the effect of Ang II on Nox4 in podocytes, we measured Nox4 protein expression with western blotting and real-time PCR analysis. Density values for Nox4 protein of the representative immunoblots from each group showed a marked increase in Nox4 protein level in Ang II-treated podocytes by 46.9 % at 24 hours after incubation (n = 4) (Fig. 3B; p < 0.01). However, probucol suppressed the Nox4 protein level significantly in both control and Ang II-treated podocytes (Fig. 3B; p < 0.05 and p < 0.01, respectively). Additionally, real-time PCR analysis was conducted for Nox4 expression (Fig. 3C). As expected, Nox4 expression was enhanced in Ang II-treated podocytes in a time-dependent manner compared with that in untreated cells (n = 4) (Fig. 3C; p < 0.05). These results suggest that a transcriptional mechanism contributes to the increase in Nox4 protein in Ang II-treated podocytes.
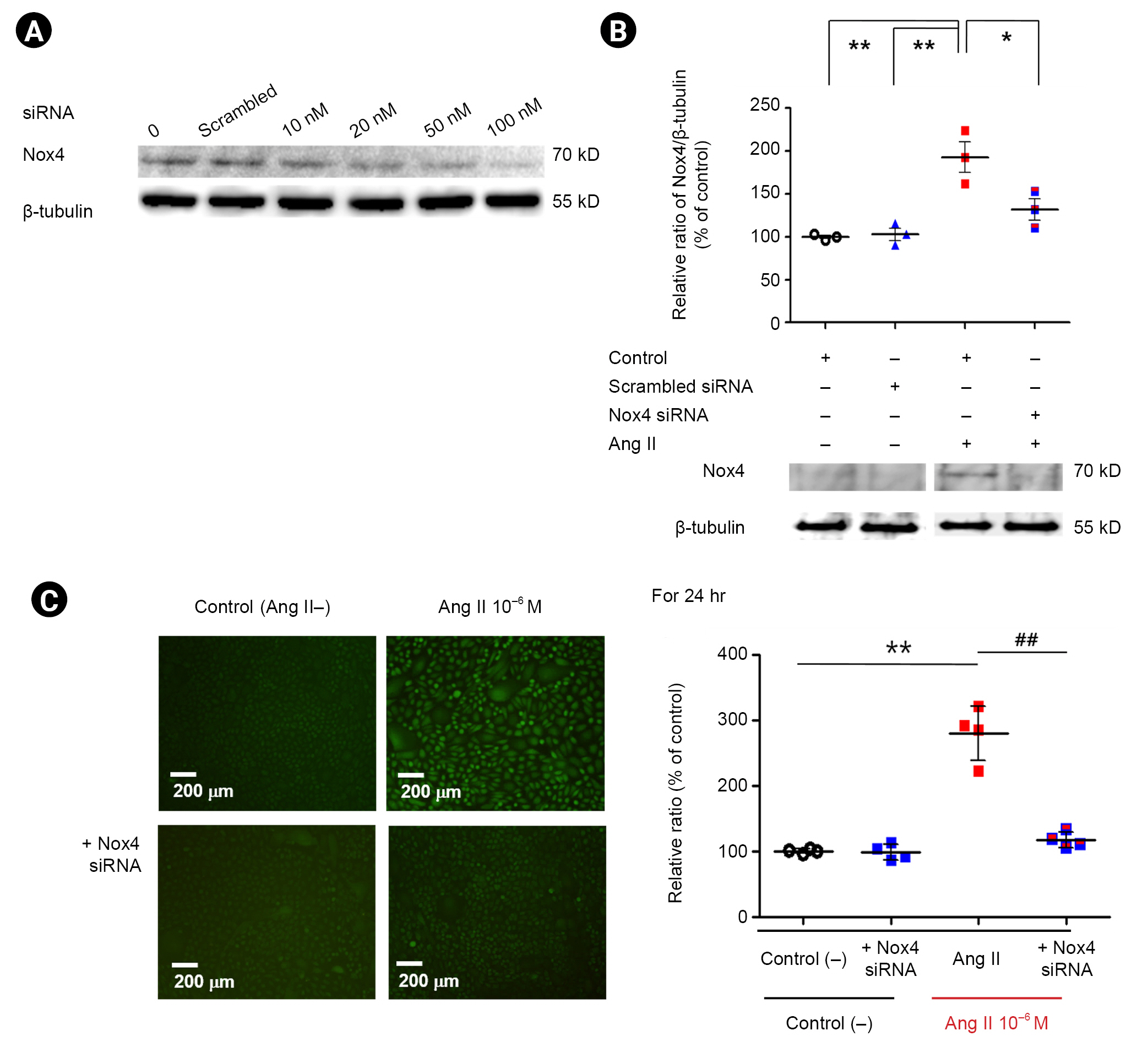
Nox4 siRNA inhibits Nox4 protein and subsequent angiotensin II-induced oxidative stress
To evaluate the changes in Nox4 protein and Ang II-induced oxidative stress caused by Nox4 siRNA, we transfected podocytes with Nox4 siRNA. We found that 50 nM of Nox4 siRNA inhibited Nox4 protein by 86% for 24 hours (Fig. 4A). As expected, Nox4 siRNA (50 nM) significantly reduced the Nox4 protein level increased by Ang II (n = 3) (Fig. 4B; p < 0.05). Additionally, Nox4 siRNA (50 nM) significantly reduced the oxidative stress increased by Ang II (n = 4) (Fig. 4C). These results suggest that Ang II promotes podocyte oxidative stress by upregulation of Nox4 in a transcriptional mechanism, and selective inhibition of Nox4 could reduce podocyte oxidative stress in Ang II-treated podocytes.
Angiotensin II induces podocyte apoptosis via oxidative stress and Nox4
We previously found that Ang II-induced podocyte apoptosis in a time- and concentration-dependent manner [5]. In this study, FACS analysis showed that treatment with 10ŌĆō6 M of Ang II increased apoptosis significantly (n = 3) (Fig. 5A and B; p < 0.01, both), but that increase was mitigated by probucol and Nox4 siRNA (n = 3) (Fig. 5A and B, respectively; p < 0.05, both). These data suggest that Ang II induces podocyte apoptosis via oxidative stress and upregulation of Nox4 that were reduced by application of an antioxidant or selective inhibition of Nox4.
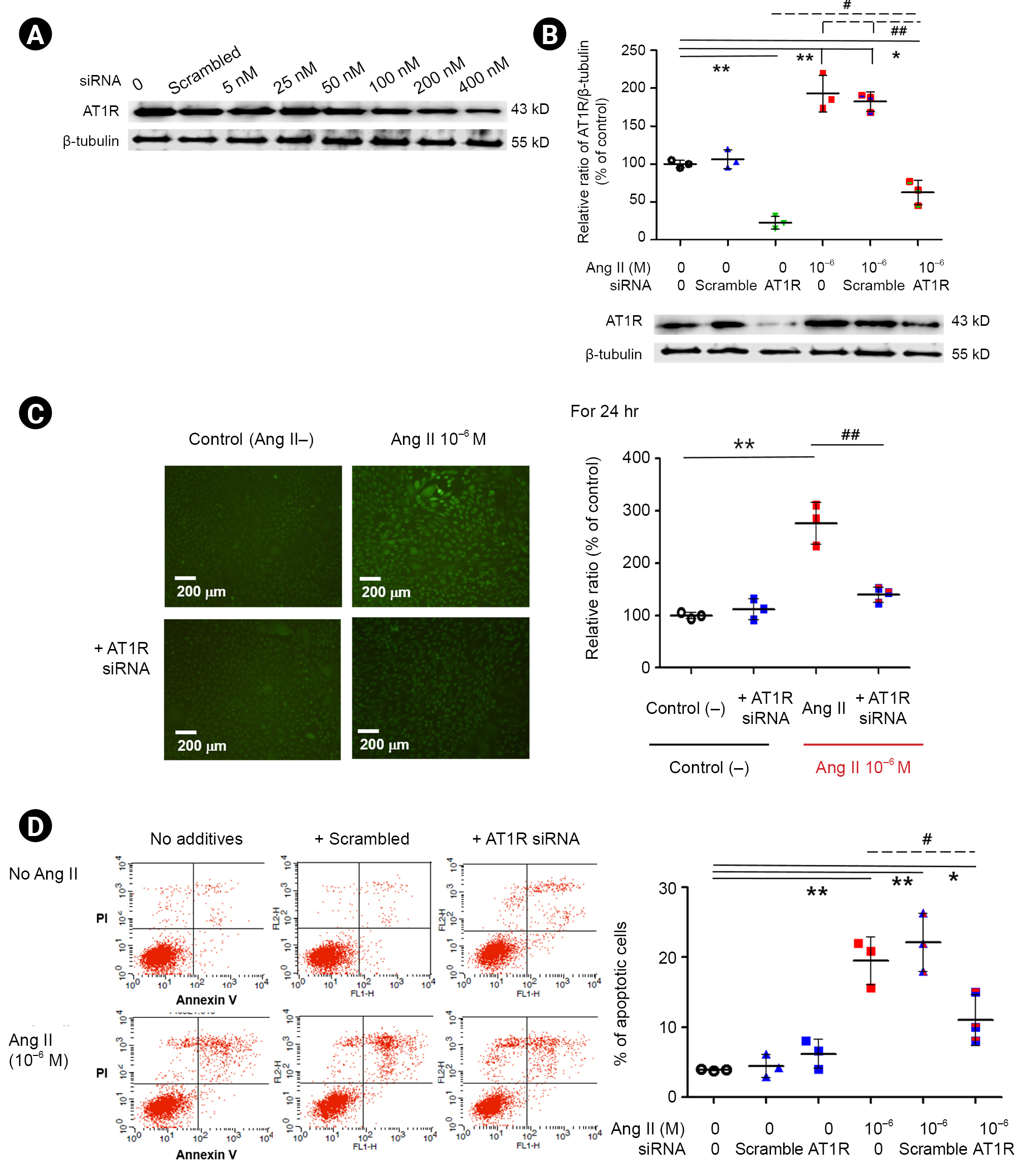
AT1R siRNA inhibits angiotensin II-induced oxidative stress and podocyte apoptosis
To evaluate the role of AT1R in Ang II-induced oxidative stress and apoptosis, we transfected podocytes with AT1R siRNA. We found that 400 nM of AT1R siRNA inhibited AT1R protein by 79% for 24 hours (Fig. 6A), and that Ang II treatment upregulated AT1R protein, which was mitigated by AT1R siRNA (n = 3) (Fig. 6B). Ang II treatment increased oxidative stress and apoptosis as in the previous results (n = 3) (Fig. 6C and D, respectively; p < 0.01, both) that were recovered by AT1R siRNA (n = 3) (Fig. 6C and D, respectively; p < 0.01 and p < 0.05). These data suggest that Ang II induces podocyte oxidative stress and further apoptosis via AT1R and that antagonizing AT1R by adding it to Nox4 inhibition could have a better outcome.
Discussion
Ang II mediates podocyte injury via hemodynamic and non-hemodynamic actions, including oxidative stress, ERS, apoptosis, and inflammation, contributing to proteinuria and glomerulosclerosis [1ŌĆō5,23]. Severe injuries cause apoptosis of terminally differentiated podocytes, which has been demonstrated in several in vitro and in vivo models [4,5,11,24ŌĆō26]. Similar to previous reports, we found that Ang II-induced podocyte apoptosis in vitro [5].
As a non-hemodynamic action of Ang II, induction of oxidative stress contributes to early podocyte ERS and apoptosis and later causes progressive renal injury [23,27,28]. Oxidative stress refers to the imbalance between generation and scavenging of ROS, such as superoxide anion and hydrogen peroxide. The release of free radicals from podocytes may contribute to glomerular injury and proteinuria. Among ROS, superoxide anion, metabolized by SODs, is not only a primary free radical generated by reduction of oxygen but also a source of other oxygen-centered radicals, such as hydrogen peroxide and hydroxyl radical, which participate in lipid peroxidation and induce cellular membrane damage [29]. Ang II decreased the cellular levels of total SOD and induced subsequent podocyte death in a concentration-dependent manner. [30]. In the present study, we determined whether Ang II promoted oxidative stress in vitro in podocytes and whether such oxidative stress increased podocyte apoptosis.
Oxidative stress in renal cells, including podocytes, may result from activation of Nox [10,11]. Ang II-induced upregulation of renal Nox and increased ROS production may participate in the development of hypertension and renal injury [11,27,31,32]. In this study, the suppressed activity of SOD increased Nox4 expression and caused a subsequent increase of >60% in superoxide anion and >2.5 times increase in the intracellular ROS levels in Ang II-treated podocytes at 24 hours. A MitoSOX index was increased and 8-oxod-G was highly accumulated under treatment with Ang II, i.e., Ang II-induced the generation of mitochondrial ROS in cultured podocytes. These findings are significant in that we demonstrated the direct effects of Ang II on the generation of mitochondrial ROS in podocytes and found that increased oxidative stress is related to increased podocyte apoptosis. Moreover, administration of probucol, an antioxidant, decreased Nox4 expression and subsequent oxidative stress and podocyte apoptosis, re-enforcing the role of Ang II in generation of ROS and subsequent podocyte apoptosis.
Ang II mediates oxidative stress and apoptosis via AT1R in the kidney [2,3]. In this study, we found that Ang II treatment upregulated AT1R protein, and inhibition of AT1R by siRNA reduced Ang II-induced oxidative stress and podocyte apoptosis. The reduction of podocyte apoptosis by AT1R siRNA in this study supports our previous findings [5], and the prevention of this by an AT1R antagonist, losartan in that case, were also reported recently [11]. These results suggest that Ang II induces podocyte oxidative stress and apoptosis through AT1R and that AT1R inhibition by siRNA and/or antagonizing AT1R could reduce podocyte oxidative stress and apoptosis.
Oxidative stress is considered an initiator and a major contributor to both ERS and autophagy/apoptosis imbalance [33]. In an aldosterone/mineralocorticoid receptor-induced podocyte injury model, ROS driven by aldosterone caused ERS and CHOP-dependent apoptosis and autophagy in cultured podocytes [34]. We previously found that Ang II could induce podocyte ERS of the PERK-eIF2╬▒-ATF4 axis via the PI3-kinase pathway, which was ameliorated by losartan, an AT1R antagonist [23]. Therefore, we aim to understand the role of the ERS pathway in Ang II-induced oxidative stress and subsequent apoptosis to augment the therapeutic potency.
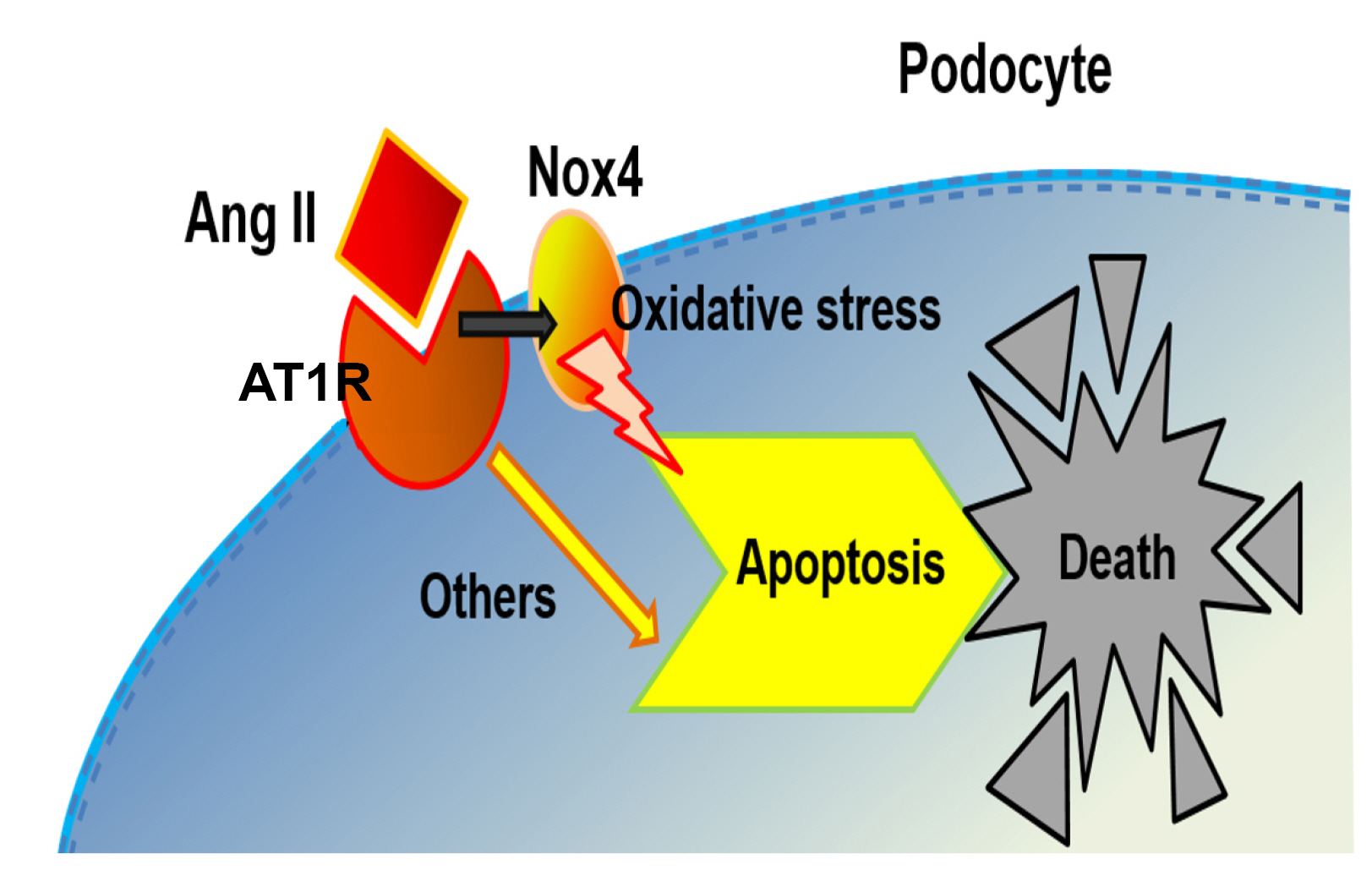
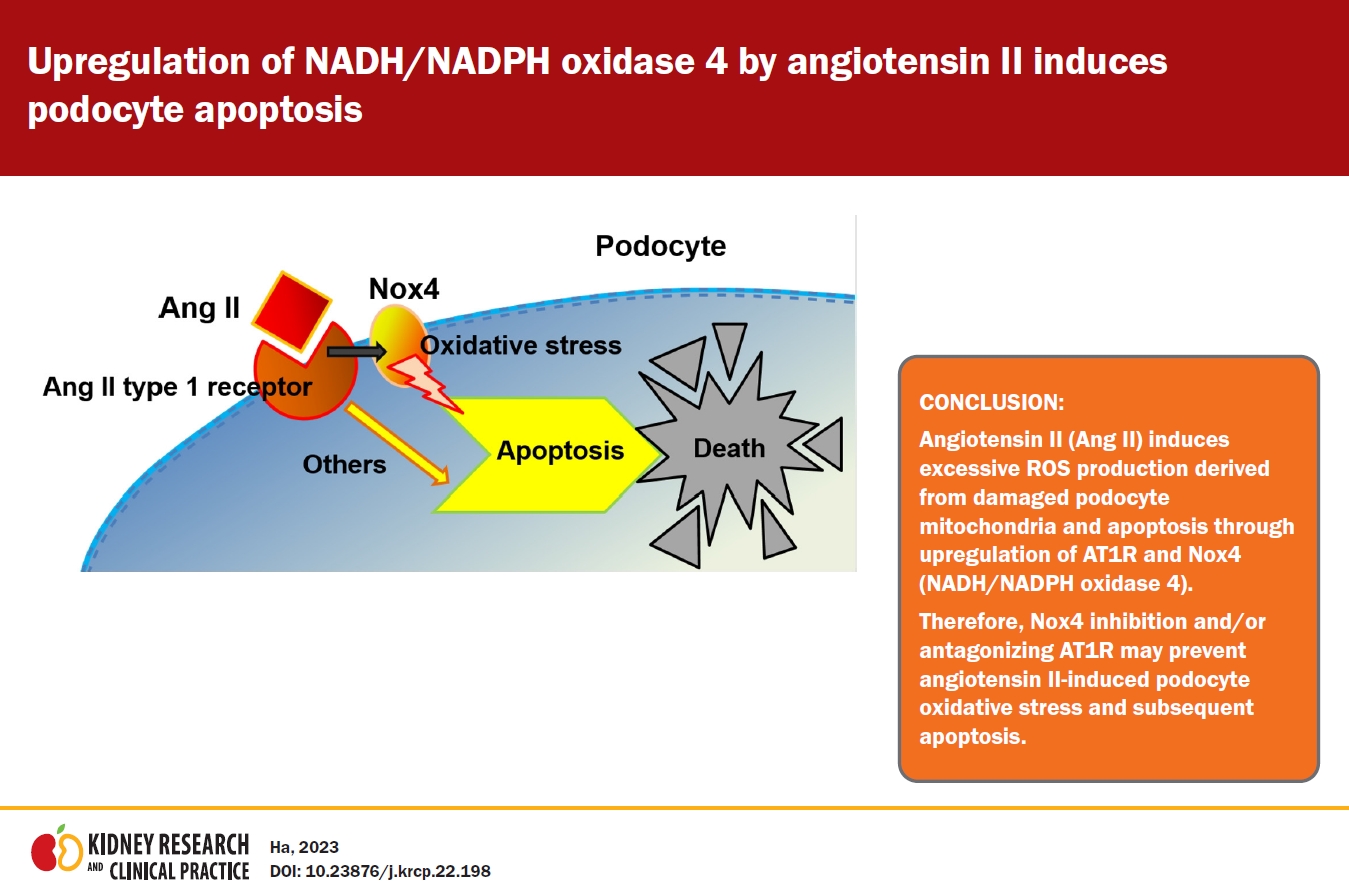
In conclusion, the cumulative data of oxidative stress and apoptosis indicate that Ang II induces excessive ROS production derived from damaged podocyte mitochondria and apoptosis through upregulation of AT1R and Nox4 (Fig. 7). Therefore, Ang II-induced podocyte oxidative stress and subsequent apoptosis through AT1R and Nox4 could be prevented by Nox4 inhibition and/or antagonizing AT1R.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









