A new pathological perspective on thrombotic microangiopathy
Article information

Abstract
Thrombotic microangiopathy (TMA) refers to a condition caused by microvascular injury that includes thrombosis, hemolytic anemia, and thrombocytopenia. There are two classic TMAs, hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura, as well as an atypical HUS (aHUS). aHUS includes a broad spectrum of disorders with diverse etiologies and shares clinical manifestations with classic TMA; however, it frequently lacks typical clinical and laboratory findings. These traits can confuse clinicians and pathologists in terms of renal pathologic diagnosis, especially in cases where TMA is associated with other glomerulopathies or hypertensive renal disease. In this review, new paradigms for classifying TMA and the diversity of histopathologic changes including associated renal diseases are discussed. Renal biopsy is an important and useful diagnostic tool for diagnosing TMA and identifying TMA changes in other renal diseases, including hypertension. Adopting the term “TMA features” for TMA-like changes in glomerulus or artery/arteriole in addition to the pathological diagnosis of glomerulopathy would be informative to clinicians for a prompt diagnosis and treatment of aHUS.
Introduction
Thrombotic microangiopathy (TMA) is a pathologic term used to describe a condition characterized by microvascular changes, including thrombosis, in association with laboratory abnormalities of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and organ injury. It is not limited to thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Severe/malignant hypertension, preeclampsia/eclampsia, antiphospholipid antibody syndrome, scleroderma, various drug toxicities, and metabolic disorders all share pathologic findings with TMA [1]. This diversity of TMA conditions has contributed to the evolution of classification criteria, which has caused some confusion regarding the diagnosis and treatment of TMA.
The TMA nomenclature is challenging in practical renal biopsy work in cases with nonthrombotic TMA features because TMA has been used to describe pathologic features of HUS and TTP, in which thrombosis in the glomerular capillary lumen and in renal arteries/arterioles was among the main findings [1,2]. Currently, such findings are rare because biopsies in such cases are very limited. Instead, the majority of biopsies are from atypical HUS (aHUS) cases or uncharacterized syndromes from HUS or TTP. Such biopsies frequently show acute and chronic microangiopathy findings but rarely show thrombi [3]. These TMA-like vascular changes are also observed in various renal diseases and have become independent findings for worse prognosis, especially in hypertension and immunoglobulin A nephropathy (IgAN) [4]. However, in the absence of thrombosis in glomeruli or vessels, such changes are not easily accepted for describing TMA.
This review discusses the evolving classification, case-based histopathologic findings, and new perspectives regarding TMA pathology, especially those associated with hypertension and IgAN.
Brief overview of the classification
The classification of TMAs is constantly evolving. Historical classifications have been based on clinical findings: TTP for predominant neurologic involvement and HUS for kidney-symptom-dominant disease. aHUS has been loosely applied to all other TMAs with no typical TTP or HUS [1,2].
By etiology, TTP is characterized by severe ADAMTS13 deficiency, while HUS is characterized by the presence of Shiga toxin-producing bacteria, and aHUS is broadly used to refer to all other causes of TMA [5].
Following the discovery of the role of complement dysregulation [1,6], a proportion of aHUS specifically refers to complement-mediated TMA or primary TMA. The term secondary TMA refers to any aHUS with an unclear cause. At least 50% of aHUS cases occur because of genetic abnormalities leading to dysregulation of the alternative complement pathway on the surface of the vascular endothelium [3,5–8]. Complement factor I and membrane cofactor protein mutations are more common in adults and children, respectively [9]. Patients with a complement factor H (CFH) mutation have reduced ESRD-free survival, whereas patients with a membrane cofactor protein mutation have longer end-stage renal disease (ESRD)-free survival [9]. However, these abnormal complement genes have been identified in patients with secondary TMA associated with infections, pregnancy, transplantation, glomerular diseases, and malignant hypertension. There is overlap between primary and secondary TMA. Of 66 Korean patients with aHUS, 36 (45%) had at least one aHUS-related variant gene. The most frequently affected gene was CFH (32%), followed by thrombomodulin gene (20%) and CD46 (18%) [10].
With this knowledge of genetic mutations, eculizumab, which is a humanized monoclonal antibody against C5, has been shown to be effective in treating patients with aHUS [11–15]. Prior to effective treatment, disease outcomes were poor, and the recommended management was frequent plasma exchange and infusion. With increasing reports of successful treatment of patients with aHUS using eculizumab, eculizumab-responsive and eculizumab-resistant aHUS have been identified [5].
Histologically, from renal biopsy, TMA can be divided into acute and chronic forms. The acute form is characterized by thrombosis in the glomerulus and/or artery or arteriole, whereas thrombi are very rare in the chronic form. When present, thrombosis is usually limited to the artery and arteriole and commonly is seen in secondary TMA forms. It has been frequently observed that the secondary form has no typical laboratory findings or evidence of hemolysis and thrombocytopenia. For these reasons, “laboratory TMA” and “morphological TMA” have become the suggested nomenclature for classification.
Based only on renal biopsy findings, the author, who is a pathologist, would like to propose the term “TMA features with or without thrombosis” in addition to the original renal diseases, because renal function deteriorates rapidly in most cases with these features [5]. Using this proposed term in biopsy reports would be highly informative for clinicians to determine the etiology of TMA, including genetic tests, and to provide early treatment to patients with TMA by applying plasma exchange or administering eculizumab.
Pathologic features of thrombotic microangiopathy
TMA exhibits pathological features that represent tissue responses to endothelial injury and is divided into acute/active and chronic changes. These changes often coexist in the same biopsy tissue.
Details of the histopathologic findings have been well defined and established [3,15]. Acute/active microangiopathy has been defined as the presence of thrombi (Fig. 1), endothelial swelling or denudation, mesangiolysis, or microaneurysms in the glomeruli; thrombi, endothelial swelling or denudation, intramural fibrin, or intimal swelling in the arterioles; and thrombi or myxoid intimal swelling in the arteries (Fig. 2A, B).

Glomerulus with capillary thrombi.
(A) Capillary thrombi (arrow, red-orange color) are confined in the capillary lumen (Masson’s trichrome stain, ×400). (B) Anti-CD61 positive clumps (arrow) are platelet aggregates and represent thrombi. The scattered granules in the capillary lumen are isolated platelets that are not evident on routine microscopy (immunohistochemistry, ×400).

Arterial myxoid and fibrous changes.
(A) Subintimal or subendothelial edema with myxoid changes (arrow) shows a pale blue color on Masson’s trichrome stain (×400). (B) Myxoid changes with fibrosis in a larger artery (Masson’s trichrome stain, ×200). (C) Thick arterial walls with fibrosis and hyalinosis (arrow, red-orange color) (Masson’s trichrome stain, ×400). (D) Multiple laminations in the arterial wall (Periodic acid-Schiff, ×200).
Chronic microangiopathy is defined as double contours of glomerular capillary walls, observed via light microscopy; widening of the subendothelial area under electron microscope (Fig. 3); presence of fibrous intimal thickening with onion skin-like lamination in the arteries; and hyalinosis in arterioles (Fig. 2C, D).
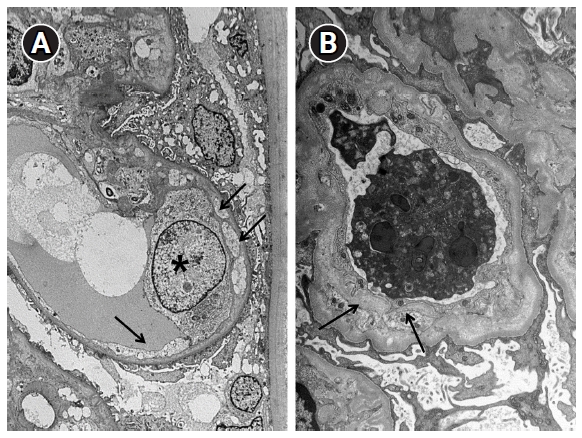
Electron microscopic changes in the glomerular basement membrane.
(A) Early changes. Endothelial cell detachment (arrows) from the glomerular basement membrane and an electron-lucent material between the detached endothelial cell and glomerular basement membrane are seen. Endothelial cell (asterisk) has edematous changes (×3,000). (B) Chronic changes. A new layer of basement membrane (arrows) forms a duplicate of the glomerular basement membrane (×5,000).
Immunofluorescence microscopy is negative for anti-immunoglobulins or complements, but fibrin clumps can be observed in glomerular capillaries and the arteriolar lumen.
Electron microscopy demonstrates glomerular endothelial cell swelling with fluffy subendothelial electron-lucent material (Fig. 3). This is visualized as a double contour of the peripheral glomerular capillary wall when observed under a light microscope. The degree of subendothelial swelling or edema varies and is not related to etiology. Intraluminal fibrin fragments occasionally can be present, but electron dense deposits are not identified.
In summary, active/acute TMA is characterized by thrombi, and glomeruli are more frequently involved than intrarenal vessels. Chronic TMA lesions are located mainly in the arteries and arterioles with or without glomerular double contours. The presence of thrombi is rare.
Because it is not easy to determine etiology from morphology, it is important for pathologists to provide a differential diagnosis, especially in patients with severe hypertension, where vascular changes are similar to TMA. Because TMA features without thrombosis can coexist with other glomerulopathies or glomerular diseases, pathologists should give this information to clinicians to rule out the etiology of aHUS and to avoid poorer renal outcomes. Practically, routine light microscopic stains for renal biopsy are not sufficient to differentiate and confirm TMA diagnosis. Therefore, several helpful ancillary tests have been proposed.
The thrombi, when present, appear as clumps of red and white blood cells and are identified by hematoxylin and eosin and other stains. These can be confirmed by CD61 immunohistochemistry, an antiplatelet antibody [16] (Fig. 1B). However, this staining is sometimes inconclusive because of nonspecific scattered fine granular positivity in the capillary lumen and interstitium.
Specific and sensitive markers of complement activation are not known for TMA. Noris et al. [17] reported strong immunostaining for C3 and C9 neoantigens in small arterioles. A Portuguese cohort study found that 17% (21/126) of IgAN cases were positive for C4d at the arteries/arterioles and were associated with hypertension and the chronic form of TMA [18]. Chua et al. [19] demonstrated the usefulness of C4d deposition in the renal tissue of patients with IgAN and TMA. C4d immunohistochemistry/immunofluorescence has become an important ancillary form of staining for antibody-mediated rejection at the peritubular capillaries [20]. It has been reported to be positive along the glomerular capillary wall in membranous nephropathy cases [21]. Several studies have shown that glomerular C4d is a valuable biomarker associated with disease progression in IgAN [22–24]. C4d immunohistochemistry performed for IgAN, membranous nephropathy, and transplant renal biopsy revealed that C4d was frequently positive in the thick arteriolar wall and the foci of arterial hyaline change (Fig. 4). Laskin et al. [25] reported that diffuse or focal renal arteriolar C4d staining was more common in cases with hematopoietic stem-cell transplantation-associated TMA (75%) compared with controls (8%). They suggested this arteriolar C4d deposition implicates localized complement fixation and causes secondary kidney injury. In malignant nephrosclerosis, renal biopsy showed identical histopathological and ultrastructural features to TMA, while arteriolar C4d was associated with deposition of C5b-9, C3a, and C5a [26].
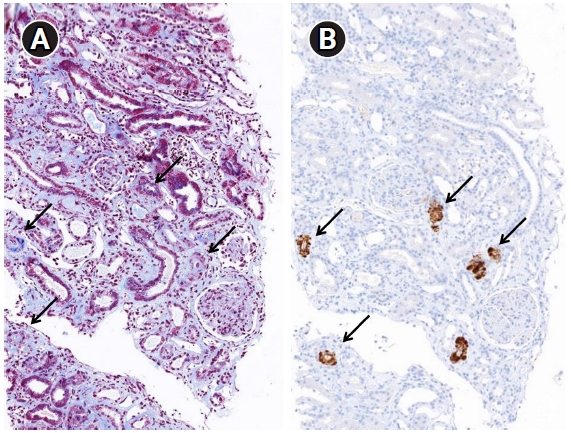
C4d depositions in arteriolar walls.
(A) Thick arteriolar wall (arrows) (Masson’s trichrome stain, ×100). (B) C4d deposition along the thick arteriolar wall (arrows) (immunohistochemistry, ×100).
Thrombotic microangiopathy associated with immunoglobulin A nephropathy
IgAN has long been recognized in intrarenal vascular lesions, such as arteriolar wall thickening and hyaline changes. This is similar to TMA and can be a prominent feature [27,28]. However, the Oxford classification does not consider these vascular changes because of their poor reproducibility and lack of statistically significant association with decreased renal survival [29,30]. Recently, Moriyama [23] and Zhang et al. [31] demonstrated that patients with arterial or arteriolar sclerosis had significantly more severe histological findings, decreased renal function, and lower survival rates than those without sclerosis (64.0% vs. 84.7%, p < 0.001). They also showed that treatment with renin-angiotensin system inhibitors improved the survival of patients with arterial or arteriolar sclerosis [23,31].
The main studies about TMA in patients with IgAN are shown in Table 1. Discrepancies in the frequency of TMA involvement might be due to differences in the definition of morphologic criteria, but differences among populations also might be relevant and included reports from the United States [26,32], Iran [33], France [16], China [31,34], Netherlands [19], Brazil [35], and Korea (Kyungpook National University Hospital [KNUH]). Most reports were based on light microscopic changes only, but two studies [26,36] and our results (KNUH) included electron microscopic changes. Zhang et al. [31] reported that, of 1,683 patients with IgAN, only 43 (2.6%) had arteriolar thrombosis. Haas and Mirocha [36] reported two cases of systemic TMA.

Studies on TMA lesions in patients with IgAN
Renal pathology findings of the reported cases in Table 1 were mostly confined to the renal arteries and arterioles. When electron microscopic examination was available, widening a subendothelial zone of the glomerulus was described. In addition to these changes, Chang et al. [32] found that all cases showed a remarkable degree of interstitial fibrosis and tubular atrophy, with interstitial inflammation.
The precise pathogenic mechanism underlying TMA in IgAN cases remains unclear. Hypertension deserves specific attention here, given that high shear stress induced by hypertension can cause microangiopathic changes [37]. However, hypertension seems unlikely to be the sole cause of IgAN-associated TMA, because the Brazilian cohort study [35] showed that 20 of 69 cases (29%) occurred in patients with systolic pressure values of <140 mmHg at the time of biopsy. Thus, antiendothelial cell antibodies were proposed as a possible pathologic contributor rather than pressure-induced endothelial disruption [38].
In a TMA study of IgAN, most laboratory findings for TMA were negative (Table 1). However, eight cases in a study by El Karoui et al. [16] and two cases in a study by Haas and Mirocha [36] showed relevant results for TMA. The group with TMA morphology who had laboratory signs of TMA had far worse renal outcomes than a group with morphologic TMA and no laboratory signs. Cases with only morphologic TMA were described by Haas and Mirocha [36] as the “underwater” part of an iceberg.
Regarding thrombocytopenia as a signature sign of HUS, a French cohort study [39] found that HUS patients with normal platelet count were not uncommon (13%), and they referred to them as partial HUS cases. Cases of HUS that did not show thrombocytopenia had similar characteristics and identically poor renal outcomes to cases with classic HUS and should be treated the same. Therefore, thrombocytopenia might not be among the main findings among HUS cases.
Genetic mutations should be considered as part of the IgAN-TMA pathogenesis mechanism, but two studies of complement mutation genes on IgAN did not identify mutations [16,40]. Currently, genetic variants of complement genes do not appear to play a role in TMA in IgAN. However, complement activation is an important trigger factor for IgAN development via lectin and alternative pathways [41,42]. The presence of the complement cleavage product C4d has been shown to be associated with progression of renal damage in patients with IgAN [22–24]. However, these results focused on glomerular lesions and not on arterial/arteriolar changes. Some case reports of gene mutations in IgAN patients were concurrent systemic TMA in IgAN cases rather than IgAN-associated TMA cases [43–46]. However, future work must address the impact of complement activation on the pathogenesis of TMA lesions in patients with IgAN.
In addition to differences in TMA incidence, common results from the above published reports (Table 1) were as follows: 1) IgAN with TMA-like arterial and arteriolar lesions had worse renal function and worse outcomes than cases without such changes; 2) the studies had no clinical or laboratory findings of TMA; 3) genetic mutations relevant to TMA were absent; and 4) the incidence of hypertension was higher than IgAN without TMA-like vascular lesions.
Taken together, these findings indicate that TMA-like pathologic features of arterial and arteriolar lesions in IgAN are TMA variants. In line with pathology diagnoses, these vascular changes should be included in the Oxford classification for worse prognostic signs, as well as for indicating the possibility of systemic TMA. Such diagnoses would be helpful for clinicians to appropriately treat patients.
Different types of glomerulopathies other than IgAN complicated by TMA have been reported, such as lupus nephritis [47], focal segmental glomerulosclerosis [48], membranous nephropathy, and antineutrophil cytoplasmic autoantibodies-associated crescentic glomerulonephritis [4,49–50]. Several mechanisms have been suggested, such as nephrotic-range proteinuria and gene mutations involving alternative complement regulatory proteins [50]. Although most reported cases were concurrent TMA with underlying glomerulopathies, C3 glomerulonephritis might be an exception due to abnormalities of the alternative pathway of complement. Approximately 5% of C3 glomerulonephritis had histopathologic features of TMA [51].
Hypertension
Long-standing uncontrolled hypertension can induce slowly progressive renal failure due to hypertensive nephrosclerosis. However, quite often, a more acute and potentially life-threatening TMA can occur with severe hypertension [52]. There has been a debate regarding whether hypertension is the cause or effect of TMA by sheer stress-induced endothelial damage [27]. However, a recent study by Timmerman et al. [53,54] identified underlying mutations in the complement genes of severe hypertension cases. In six of nine patients, they found C3 mutations in three, CFI in one, CD46 in one, and CFH in one patient. In contrast to patients without genetic defects, patients with complement defects invariably progressed to ESRD, and disease recurrence after kidney transplantation was common. Timmerman et al. [53,54] showed that some patients with hypertension-associated TMA should be considered in the spectrum of complement-mediated TMA, the prognosis of which is poor even though profound hematologic symptoms of TMA were uncommon. Hence, the authors proposed testing for genetic complement abnormalities in patients with severe hypertension [53,54]. Renal biopsy findings in available cases were vascular TMA rather than glomerular changes. Prominent myxoid intimal change and onion-skinning were described.
TMA changes within the renal vasculature can induce severe hypertension, and vice versa. Thus, identification of aHUS in patients with severe hypertension could be challenging, particularly in patients without systemic hemolysis signs [53], which yields TMA laboratory signs, including anemia, schistocytosis, thrombocytopenia, and elevated lactate dehydrogenase. For suspected TMA cases with hypertension, a confirmatory kidney biopsy can be the first step to detecting TMA.
These vascular TMA morphologies with no systemic TMA signs have also been seen in pregnancy-related TMA [55,56] and transplant glomerulopathy in allografts [57–59]. Light microscopy revealed arterial/arteriolar changes and electron microscopy revealed subendothelial edema in the glomerular basement membrane. In addition to a slow but persistent progressive clinical course, the term “chronic smoldering TMA” was suggested [55,57–59].
Conclusion
TMA is a heterogeneous disorder induced by endothelial injury with many etiologies. With the exception of two prototypes of HUS and TTP, aHUS does not have the typical clinical signs or pathologic renal changes of TMA. aHUS frequently lacks thrombocytopenia and MAHA. Moreover, aHUS shows the presence of thrombi in glomeruli or intrarenal vessels. Organ symptoms are usually limited to the kidney. Thus, many terms have been proposed to describe these atypical forms of HUS. Morphologic TMA is used in cases lacking clinical or laboratory signs. Vascular TMA is used if the disease is limited to renal arterioles/arteries. Smoldering TMA is used to describe mild but chronic and persistent clinical progression. Through this review, we show that IgAN and hypertension have vascular TMA changes frequently result in worse prognosis than patients without such changes. In the absence of thrombi in renal biopsy, TMA can be overlooked and underappreciated, even in various renal diseases. Although complement system dysregulation and genetic mutations were shown to be etiologies or triggers for TMA, routine complement tests and markers of complement activation are not suitable for prompt diagnosis of TMA because they lack sensitivity and specificity, are time-consuming, and are expensive.
In conclusion, renal biopsy is important and can be a useful diagnostic tool to diagnose TMA as well as to find TMA changes in other glomerulopathies or renal diseases including hypertension. These pathological changes and TMA features should be further described for pathologic diagnosis of glomerulopathy. Using the term “TMA features” in biopsy reports would be very helpful for clinicians to determine the etiology of aHUS, including genetic tests, and can allow early treatment to patients with aHUS by applying plasma exchange or administering eculizumab.
Notes
Conflicts of interest
The author has no conflicts of interest to declare.
Acknowledgements
I would like to thank Dr. Man Hoon Han and Dr. Mee-Seon Kim for their work in the Department of Pathology and reviewing the renal cases and supporting this study.