Pathomechanism of oxidative stress in cardiovascular-renal remodeling and therapeutic strategies
Article information

Abstract
The high prevalence of cardiovascular disease in patients with chronic kidney disease indicates significant interactions between pathogenic pathways operating in the kidney and heart. These interactions involve all cell types (endothelial cells, smooth muscle cells, macrophages, and others), components of the vasculature, glomeruli, and heart that are susceptible to oxidative damage and structural alterations. A vicious cycle occurs whereby harmful factors such as reactive oxygen species and inflammation damage of vascular structures that themselves become sources of additional dangerous/toxic components released into the local environment. The evidence of this vicious cycle in chronic kidney disease should therefore lead to add other factors to both traditional and nontraditional risk factors. This review will examine the processes occurring during progressive kidney dysfunction with regard to vascular injury, renal remodeling, cardiac hypertrophy, and the transversal role of oxidative stress in the development of these complications.
Introduction
The development of cardiovascular-renal remodeling in chronic kidney disease (CKD) is a progressive, long-term process leading to increased morbidity and mortality [1]. According to a recent analysis of the global burden of CKD, the prevalence of cardiovascular disease (CVD) accounted for an additional 1.4 million deaths among patients with CKD, which represented 7.6% of deaths due to CVD. CVD is therefore the leading cause of death among end-stage renal disease (ESRD) patients undergoing dialysis [2,3]. The interplay between kidney dysfunction and CVD is associated with both traditional atherosclerotic and nontraditional cardiovascular risk factors, including prothrombotic and proinflammatory states, endothelial dysfunction, and oxidative stress [4]. Therefore, to shed light on the pathological mechanisms underlying CVD and renal remodeling, this review will specifically address alterations in the vasculature during progressive kidney dysfunction.
The vicious cycle
It is well established that the traditional risk factors for CVD in CKD patients are hypertension, diabetes, dyslipidemia, and smoking [5–8]. However, compelling evidence also indicates pivotal roles for additional nontraditional risk factors, including inflammation, oxidative stress, and endothelial dysfunction, in the development of CVD [4,9]. When acting together, these processes encompass more than the simple concepts of traditional and nontraditional risk factors and create a vicious cycle that exacerbates hypertension, vasomotion, and vascular remodeling, leading to increased peripheral resistance, arterial stiffness, endothelial dysfunction, and atherosclerosis [4,9]. As an example, the very close relationship between oxidative stress and inflammation determines the risk of progressive atherosclerotic CVD in CKD and dialysis patients. In this context, the endothelial damage caused by free radicals creates a positive feedback loop to induce further formation of those species. During endothelial lipid peroxidation, the release of a cytotoxic end product (malondialdehyde) is pivotal to the onset of atherosclerosis and in the endothelium-dependent vasodilation and nitric oxide (NO) pathway [10]. Further, the endothelium itself plays an important role in the vasomotion of smooth muscle cells involving a complex signaling, which includes calcium oscillation, potassium efflux, NO, and cyclic guanosine monophosphate in order to induce vasoconstriction and peripheral resistance [11]. Variations in the perfusion pressure in the kidneys are detected by intrarenal baroreceptors, which modulate the renin-angiotensin-aldosterone system (RAAS). There is evidence that injured kidneys have aberrant sympathetic nerve activity which overrides the negative feedback loop on efferent renal sympathetic nerve activity thereby resulting in chronically increased systemic blood pressure [12]. In addition, hypertension causes tubulointerstitial fibrosis, which is associated with reduced secretory solute clearance, while reduced estimated glomerular filtration rate (eGFR) is linked to greater left ventricular wall thickness that predisposes patients to CVD [13].
Although an association between hypertension and inflammation is established, it is still unclear whether inflammation is predominately a cause or an effect of hypertension. Hypertensive patients are reported to have high plasma C-reactive protein (CRP) concentrations, and elevated plasma CRP concentrations in prehypertensive subjects are associated with a greater risk of developing overt hypertension [14]. Systemic low-grade inflammation, as assessed by CRP and other inflammatory markers such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, is also able to upregulate the angiotensin type 1 receptor (AT1R), the angiotensin II (Ang II) receptor mainly involved in the production of oxidative stress species and in augmenting AngII signaling [15].
All the molecular mechanisms that link oxidative stress, inflammation, and hypertension have also been shown to be involved in the pathogenesis of atherosclerosis and subsequent renal remodeling.
Atherosclerotic injury
The early stages of atherosclerosis are thought to begin with endothelial injury and inflammatory processes where cytokines and adhesion molecules are recruited not only as components of the acute phase response but also of a more chronic response that further stimulates innate and adaptive immune responses [16]. The very early stages of atherosclerosis entail accumulation of low-density lipoproteins (LDLs) within the arterial walls of vessels prone to oxidation or modification that are particularly prevalent in subjects with CKD. The oxidation of LDL (oxLDL) itself is a crucial event in the atherogenesis that is particularly evident in patients with ESRD and undergoing dialysis [17]. In addition, during atherosclerotic injury, the endothelium modifies its functionality by increasing the expression of cell adhesion molecules, such as selectin, intercellular adhesion molecule 1 (ICAM-1), and the vascular cell adhesion molecule 1 (VCAM-1), which allows subsequent monocytes rolling and infiltration [16]. Through the action of specific proteins such as monocyte chemoattractant protein-1 (MCP1), IL-8, and fractalkine, monocytes can migrate through the endothelium and into the intima, where they differentiate into macrophages that take up oxLDL (Fig. 1). This ineffective clearance of cholesterol-rich lipids within the arterial wall leads to accumulation of macrophage-oxLDL complexes and the ensuing formation of proinflammatory foam cells. Excessive numbers of foam cells are present in CKD patients, whose macrophages also display activated nuclear factor kappa B (NF-κB) and reduced expression of the ATP-binding cassette transporter A1, which is responsible for energy-dependent efflux of cholesterol [18]. The NF-κB transcription factor mediates multiple aspects of innate and adaptive immune function and plays a crucial role in CVD by inducing the transcription of proinflammatory genes, by causing glomerular injury and by activating acute stress responses.
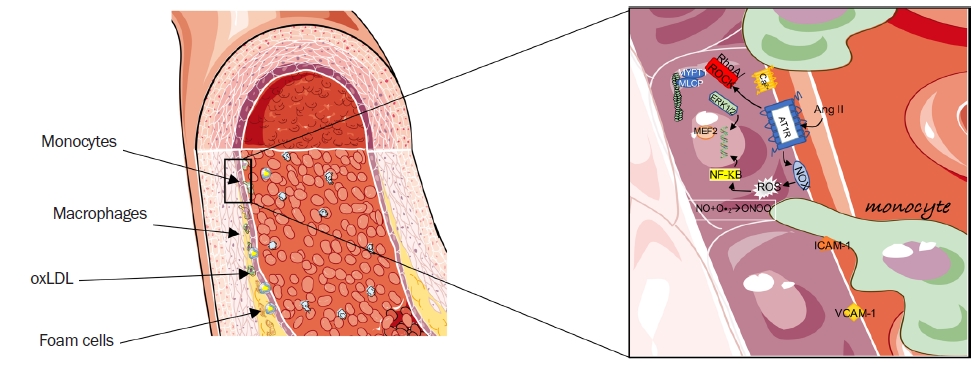
The atherosclerotic injury process.
(A) A representative image of an atherosclerotic artery. (B) High-magnification image of the initial stages of the atherosclerotic process. Monocytes infiltrate through the endothelium following increased expression of the adhesion molecules vascular cell adhesion molecule-1 and intercellular adhesion molecule-1. In parallel, intracellular signaling promoted by hormonal stimuli such as AngII triggers increased oxidative stress, vasoconstriction, and profibrotic responses. Image modified from smart.servier.com.
AngII, angiotensin II; AT1R, angiotensin receptor type 1; RhoA/ROCK, RhoA-Rho kinase; ERK 1/2, extracellular-signal-regulated kinase 1/2; ICAM-1, intercellular adhesion molecule 1; MEF2, myocyte-enhancer factor 2; MLCP, myosin light chain phosphatase; MYPT-1, myosin phosphatase target protein 1; NF-κB, nuclear factor kappa B; NO, nitric oxide; ONOO–, peroxynitrite; oxLDL, oxidation of low-density lipoprotein; VCAM-1, vascular cell adhesion molecule 1.
The presence of oxLDL can also lead to recruitment of toll-like receptors, which aggravate plaque inflammation. In the phase of atherosclerotic injury, macrophages are also recruited through specific signals transduced by the release of granules containing α-defensins, cathepsin G, and azurocidin from neutrophils [19]. Those proteins are an additional signal to activate macrophages and to initiate foam cell differentiation. Once formed, foam cells lose their motility and become trapped within the atherosclerotic plaque. The intracellular mechanisms that reduce migration capacity of macrophages may be related to the RhoA/Rho kinase (ROCK) pathway, which is also a key signaling pathway in the progression of cardiovascular remodeling. In addition, RhoA signaling regulates the stabilization of vessels during vascular development and cardiac development during embryogenesis, and modulates vascular smooth muscle cell (VSMC) contractility [20]. Cholesterol-laden macrophages appear to display reduced RhoA function, with consequently reduced ROCK activity that lowers myosin light chain phosphorylation and impairs motility of foam cells, thus promoting atherosclerotic plaque formation [21]. Conversely, inhibiting RhoA/ROCK activity regresses atherosclerotic plaques, although the mechanism of this effect is not clearly understood.
In addition, other cells are involved in the formation of atherosclerotic plaques, including T lymphocytes (subtypes Th1 and Th2) that release proinflammatory cytokines and are associated with progression of atherosclerosis for their role in the polarization of macrophages [22].
Vascular injury
The progression and development of atherosclerosis are determined by the degree of endothelial damage, which is in turn influenced by the detrimental action of reactive oxygen species (ROS) when they are not effectively scavenged by antioxidant defenses. The redox imbalance that induces oxidative stress (excess ROS and/or reduced antioxidant defensive capacity) contributes to the inflammatory state and vascular remodeling [4]. One of the most common events in kidney failure is the accumulation of uremic toxins, such as advanced glycation end products (AGEs), asymmetric dimethylarginine (ADMA), indoxyl sulfate, p-cresyl sulfate, and trimethylamine N-oxide [1]. The presence of these substances influences the synthesis or degradation of other substances produced by physiological processes worsening the already precarious state of the vasculature in CKD. For example, the accumulation of AGEs that bind to collagen and elastin induces arterial and myocardial stiffening [23]. Given that collagen and elastin are the main components of vascular walls, vascular strength and elasticity are influenced by their oxidative state and susceptibility to cross-linking. As such, AGE-linked collagen is resistant to hydrolytic turnover that reduces the strength and elasticity of vascular walls. In addition, the activation of receptors for AGE in endothelial cells, macrophages and lymphocytes reduces vasodilation, vascular permeability, mononuclear cell migration and platelet adhesion, and uptake of macrophages induced by LDL cross-linking.
Another factor also associated with vascular aging is the α-klotho enzyme that favors increment of serum phosphorus, reduced expression of which is linked to increased oxidative stress and decreased NO availability that together exacerbate endothelial dysfunction and, contribute to cardiovascular risk. In a study of 77 Chinese adult CKD patients, α-klotho expression decreased over time concurrently with reductions of kidney function, while ADMA levels increased steadily [24]. A reduction in α-klotho favors the progression of vascular calcification (in particular of arterial walls) following calcification of atherosclerotic plaques due to hyperphosphatemia. Evidence for the role of α-klotho in vascular remodeling comes from a defective α-klotho mutant animal models that showed extensive calcification in the medial layer of the aorta, medium-sized arteries, and small arteries [25]. The α-klotho protein functions as a co-receptor or scaffold protein for the fibroblast growth factor (FGF) receptor (FGFR). Binding of FGF23 produced in bone to the renal FGFR regulates phosphate reabsorption and calcitriol production in the kidney; therefore impaired FGF23 activity due to reduced α-klotho expression leads to dysregulation of phosphate and vitamin D homeostasis thereby contributing to vascular calcification [26]. Phosphatemia is also associated with increased ROS and free radicals, which can inhibit endothelial NO synthase (NOS) activity to reduce NO productionand increase peroxynitrite (ONOO–) generation [27]. Inhibition of NOS by endogenous ADMA may further contribute to endothelial dysfunction and vascular remodeling (Fig. 1). The interactions between ROS and other proteins are responsible for altered intracellular signaling resulting in impairment of contractility and elasticity of vessels.
Tubules of damaged kidneys display only residual activity of transport systems resulting in the concentration of toxins [28].
A critical event in renal remodeling and fibrosis is the loss of polarity of renal tubule endothelial cells and the subsequent transition to a mesenchymal phenotype, termed the epithelial-to-mesenchymal transition (EMT) [29]. During this process, epithelial cells lose epithelial markers such as E-cadherin (one of the main components of adherent junctions) in favor of mesenchymal markers such as alpha-smooth muscle actin (α-SMA). This transition leads to progressive loss of cell junctions and migration of mutant cells towards the interstitial space, which, after complete transition, become myofibroblasts synthesizing α-SMA and matrix proteins and collagen that contribute to tubulointerstitial fibrosis [29].
Renal remodeling
The international guidelines of the Kidney Disease Improving Global Outcomes (KDIGO) group identify CKD as the presence of a glomerular filtration rate (GFR) < 60 mL/min/1.73 m2 for at least 3 months [30]. The KDIGO guidelines also recognize a prognostic role for albuminuria associated with CVD independently of eGFR [31–33]. The reduction of the filtration rate implies that several uremic toxins are not fully excreted by the kidneys. Those substances can be categorized based on their molecular weight and are all associated with the progression of CKD and increased risk of CVD. Among them, the smallest (<500 Da), such as urea, uric acid, and ADMA, are particularly associated with inflammation; those of medium sizes (>500 Da), such as β-2 microglobulin and endothelin, have a role in increased blood pressure, oxidative stress, inflammation, endothelial dysfunction, and arterial stiffness, The largest are solutes, such as indoxyl sulfate and AGEs and other solutes of intestinal origin, bound to plasma proteins (chiefly albumin) that are associated with the onset of cardiovascular events [34,35]. In ESRD patients undergoing renal replacement therapy (hemodialysis or hemofiltration procedures), the efficiency of uremic toxin removal depends on the type of filtration membranes used, which will be discussed further below. However, in addition to involvement in the progression of kidney disease, the endothelium is subjected to inflammation and oxidative-stress-mediated damage that affect endothelial function and the generated inflammation and oxidative stress signaling spread to the surrounding structures amplifying the damage. The cross-talk between endothelial cells of the innermost side of the vessel wall and VSMCs mediates acute and chronic changes in tissue perfusion resulting from vasodilation or vasoconstriction, activation of platelet aggregation or its inhibition, and other effects. [36]. The mediators involved are mainly cytokines and growth factors that regulate changes in the proliferative and migratory status of VSMCs, endothelial function, and extracellular matrix composition. Specifically, NO, endothelin-1, FGF, and transforming growth factor beta (TGF-β) act on endothelial cells, while VSMCs are stimulated also by hormones such as AngII and epinephrine and by IL-1 and interferon-gamma. In CKD, increased circulating ROS, cytokines and the concomitantly reduced bioavailability of NO (diverted to production of peroxynitrite in the presence of ROS) promote dysfunctional VSMCs to migrate toward the intima to cause intimal hyperplasia and deposition of abnormal extracellular matrix and hyaline material, vascular calcification with stiffening of arteries and high pulse pressure [37,38]. Calcification of central vessels contributes to increased pulse wave velocity, earlier reflection of the pulse wave, and increased cardiac afterload, all of which contribute to heart failure.
Patients with CKD seem also to exhibit much greater susceptibility to the adverse renal effects of even moderate hypertension. Any increase in blood pressure within the intrarenal vasculature is of sufficient magnitude to result in barotrauma of the local vasculature. Chronic uncontrolled blood pressure leads to progressive hypertensive tubulointerstitial and glomerular nephropathy, in which the major outcome of hypertension-induced renal damage is hyperfiltration and hypertrophy of nephrons [39]. The initial damage to small vessels, where VSMCs are replaced by hyaline material, results in more expansible arteries and hemodynamic changes in the aorta. This damage later extends to the glomeruli with partial ischemia, reduced filtration, and increased oxidative stress and inflammation that further induce podocyte loss, tubule-interstitial fibrosis, and EMT [39].
Arterial remodeling and stiffness progress rapidly in CKD and hemodialysis patients, causing not only chronically elevated blood pressure but also increased blood pressure variability [4]. There is some debate as to the role of blood pressure variability in the progression of renal dysfunction; however, there is evidence that patients with CKD have a greater prevalence of sleep disorders, such as obstructive sleep apnea and restless leg syndrome, that are associated with increased blood pressure variability in CKD patients, progression of CKD, and enhanced mortality in ESRD patients [40].
Left ventricular hypertrophy and cardiac fibrosis
Cardiovascular complications are commonly fatal in patients with CKD. The main manifestation of CVD is heart failure with preserved ejection fraction, characterized by left ventricular hypertrophy (LVH) and diastolic dysfunction [41]. In the progression of kidney injury, LVH might also influence the development of heart failure with reduced ejection fractions, arrhythmias, ischemic heart disease, and sudden cardiac death. The function and regulation of the kidneys and heart are strongly intertwined such that dysfunction in one organ may induce dysfunction in the other (Fig. 2).
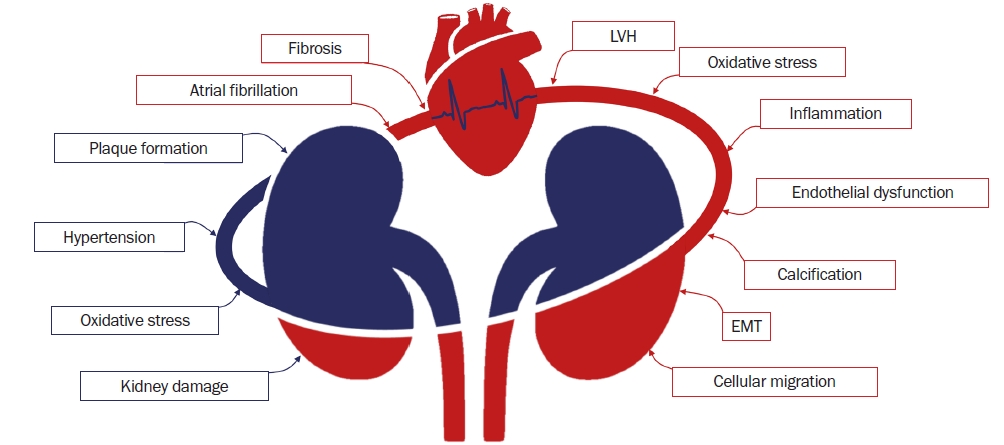
The feedback loop between kidney dysfunction, vascular injury, and heart disease.
All of the pathways involved in the progression of kidney dysfunction (e.g., reduced filtration rate and cellular migration), vascular injury (represented by the curved lines between the kidneys and the heart; e.g., endothelial dysfunction, inflammation, and plaque formation), and of heart disease (represented by the typical waves at electrocardiogram tracing; e.g., atrial fibrillation and fibrosis) are intertwined in the development of cardiovascular disease and during renal remodeling in CKD. Among these, the most common trigger factor is oxidative stress.
EMT, epithelial-to-mesenchymal transition; LVH, left ventricular hypertrophy.
There are common patterns detectable in the vasculature, glomerulus, and myocardium due to the presence of various metabolically active cell types that control the activity of peripheral structures and are susceptible to oxidative stress. For example, endothelial cells regulate vascular tone through the signaling of AngII, calcium, NO, RhoA/ROCK pathway, mitogen-activated protein kinases, TGF-β, and other pathways. Upon specific stimuli, VSMCs can alter their phenotype, developing characteristics reminiscent of osteoblasts, fibroblasts, and even macrophage-like cells with phagocytic properties [42]. Cardiomyocytes are force-producing and pacemaker cells. Therefore, cell-to-cell connections are crucial in the development of fibrosis and arrhythmogenesis, in fact, remodeling of atrial myocytes structure and electrical activity contribute greatly to its pathogenesis [43].
One of the major contributors to renal and cardiovascular fibrosis is the RAAS through the action of its major hormonal effector AngII. Further, overactivation of oxidative stress in CKD and AT1R-mediated ROS are responsible for the induction of the RhoA/ROCK pathway that drives, vasocontraction and profibrotic responses [20]. The downstream effector of ROCK, the myosin phosphatase target protein 1 (MYPT)-1, is the regulatory subunit of myosin light chain phosphatase. Inhibition of MYPT-1 phosphorylation mediated by ROCK increases myosin light-chain kinase activity with ensuing contraction of the VSMCs. Moreover, ROCK induces activation of additional genes involved in inflammation and proliferation (such as extracellular-signal-regulated kinases [ERK] 1/2) and in cytoskeletal rearrangement (through its targets ezrin, radixin, and moesin) [20]. We found that patients with stage 3 to 4 CKD displaying LVH have remarkably increased phosphorylation of MYPT-1 compared to stage 3 to 4 CKD patients without LVH. In addition, in patients with LVH, MYPT-1 phosphorylation correlated positively with left ventricular mass [44]. Blocking ROCK activity with the Rho-kinase inhibitor fasudil reduces activity in the downstream MYPT-1 pathway in a dose-dependent manner, similar to experimental animal models of AngII-induced hypertension where ROCK inhibition prevented AngII-induced LVH and myocardial fibrosis [44–46]. Upstream inhibition of ROCK using angiotensin receptor blockers reduces both p63rhoGEF (activator of RhoA) and MYPT-1 phosphorylation, providing further evidence for a pivotal role of ROCK as a switch in the progression of cardiovascular and renal remodeling [47]. As kidney injury progresses, cardiac remodeling progresses in parallel so that in patients undergoing hemodialysis treatment LVH is particularly evident. In CKD patients, LVH is correlated with markers of oxidative stress including p22phox, the subunit of nicotinamide adenine dinucleotide phosphate oxidase (NOX) essential for electron transport from heme moieties to molecular oxygen to produce superoxide, and with oxLDL [44,48]. Patients who undergo peritoneal dialysis are also subjected to progressive oxidative stress, which can become very critical to address in order to control the progression of CVD in these patients.
In studies of patients undergoing chronic dialysis, an interesting link between ROCK activity and cardiac electrical disturbances, as assessed by the relationship between MYPT-1 phosphorylation and connexin 40 (Cx40) expression, has been reported [44,49]. As an integral membrane protein of heart cell gap junctions, Cx40 is fundamental for rapid cell-cell transfer of action potentials, and increased expression of Cx40 enhances the vulnerability of the atrial myocardium to atrial fibrillation [50]. We reported that patients undergoing dialysis with permanent atrial fibrillation display increased Cx40 expression compared to dialysis patients without atrial fibrillation as well as increased MYPT-1 phosphorylation. This latter in atrial fibrillation patients also correlated with Cx40, with left atrial systolic volume, and with cardiac mass [49].
In addition, ROCK activation mediates Ca2+ sensitization that in turn regulates myocyte-enhancer factor 2-dependent expression of myocardin, which is a specific transcriptional coactivator of the serum response factor (SRF) for cardiac and smooth muscle [51]. Fibroblast differentiation into myofibroblasts is also influenced by ROCK-induced alterations in the gene expression profile of myofibroblasts and disruption of stress fiber formation and induction of myocardin-related transcription factor (MRTF) [52]. The MRTF acts in a similar manner to myocardin as a coactivator of SRF to regulate actin polymerization and the expression of α-SMA and extracellular matrix proteins, including fibronectin and collagen [20]. Taken together these mechanisms provide evidence for the critical role of ROCK in endothelial permeability, cytoskeletal rearrangement, and fibrosis.
All of the molecular mechanisms and the inter/intracellular signaling that promote fibrosis in patients with CKD also involve collagen deposition and maladaptive ventricular hypertrophy with cardiac dilation. In addition, arterial stiffness, increased systemic resistance, and systolic hypertension initially cause concentric LVH that later becomes eccentric hypertrophy, with subsequent left ventricular dilation and reduced ejection fraction due to continuous left ventricular overload. Preload-related factors also contribute to LVH and comprise the expansion of intravascular volume resulting in volume overload, extension of myocardial cell length, and eccentric or asymmetrical remodeling of the left ventricle [9].
In summary, several processes arising from excessive oxidative stress (ROS, free radical formation, reduced antioxidant defenses) and inflammation are simultaneously activated in the kidney, vasculature and heart in CKD, and act in a feedback loop to produce detrimental effects on the progression of CVD (Table 1).

Processes activated during progressive chronic kidney disease with their respective main pathways and mediators involved
Therapeutic strategies for chronic kidney disease aimed at reducing oxidative stress
Due to the complex molecular and vascular mechanisms underlying CKD, early recognition and intervention are crucial to control disease progression and to reduce morbidity and mortality in patients. Oxidative stress is one of the most important factors contributing to the development and progression of kidney disease, hypertension, and CVD, hence targeting oxidative stress might be a useful therapeutic strategy in CKD and dialysis patients.
Oxidative stress in arterial hypertension
Hypertension is closely linked with renal disease, with a prevalence that ranges from 60% to 90%, depending on the CKD stage [53]. Mechanisms such as volume overload, sympathetic overactivity, endothelial dysfunction, salt retention, hormonal alterations and increased activity of the RAAS act jointly to induce arterial hypertension; in addition, increased oxidative stress significantly and negatively influence all of these. Angiotensin-converting enzyme inhibitors (ACEi) and AngII receptor blockers (ARBs) are the first-line treatment for primary hypertension in CKD and their beneficial effects rely not only on hemodynamic/antihypertensive effects but also on anti-inflammatory/antioxidant and antifibrotic properties [54]. In this regard, we documented the efficacy of olmesartan medoxomil (ARB) treatment in hypertensive patients in the reduction of oxidative stress (as assessed by reduced p22phox, ERK 1/2, p63RhoGEF, and MYPT-1 phosphorylation and induction of antioxidant defenses such as heme-oxygenase 1 [HO-1]), calcitonin-gene-related peptide, and increased circulating endothelial progenitor cells [47].
Oxidative stress in posttransplant hypertension and electrolyte imbalances
Regulation of blood pressure is also particularly crucial for kidney transplant recipients, since hypertension is a common occurrence during treatment with calcineurin inhibitors that increase sodium retention through the increased activity of thiazide-sensitive sodium chloride cotransporter (NCC). Calcineurin blocking prevents the inhibitory effects of calcineurin on ‘with-no-lysine’ kinases (WNK), glucocorticoid-regulated kinase 1, STE20/SPS1-related proline alanine-rich kinases (SPAK), and oxidative stress-responsive protein type 1 kinase (OSR1) that are instead a switch for NCC activation [55]. Notably, it has been reported that AngII has a direct effect on sodium retention through NCC activation, further demonstrating its involvement in the induction of hypertension associated with oxidative stress [56]. If first-line antihypertensive therapy with ACEi or ARBs is not sufficient, additional therapy with diuretics, β-blockers, and calcium channel blockers can be considered [57]. Of note, in the recent update of the KDIGO guidelines for the management of blood pressure in CKD patients not receiving dialysis, particular attention has been paid to lifestyle interventions, including dietary salt restriction, physical activity, weight loss, and reduction of alcohol consumption [30].
Along with hypertension, patients with CKD and ESRD are also prone to develop metabolic disorders, especially acid-base and electrolyte imbalances [58]. Two common adverse occurrences in CKD and ESRD are hyperkalemia and hyperphosphatemia. Recurring symptoms and signs of hyperkalemia span from muscle weakness to paresthesia, paralysis, cardiac arrhythmias, and cardiac arrest [59]. The most common first-line treatment for hyperkalemia includes cellular membrane stabilization by administration of intravenous salts, and second-line treatment includes shifting potassium from the extracellular to the intracellular compartment through the administration of insulin and β-adrenergic agonists [60]. However, to clear excess potassium adequately, potassium-binding agents, dialysis, and loop diuretics are the most effective methods [60].
Another complication arising in CKD and involved in the onset and progression of CVD is hyperphosphatemia (mediated by parathyroid hormone and vitamin D), which is closely linked to bone mineral metabolism and vascular calcification as already described [58]. Management of hyperphosphatemia requires reduction of dietary phosphate intake (<1,000 mg/day) and the use of phosphate binders, which are classified into calcium- and non-calcium-based binders. However, treatment of CKD with the associated mineral bone disorder is demanding, because of the importance of managing several aspects not only related with supplementation of vitamin D but also with potential secondary hyperparathyroidism [61].
Oxidative stress in dialysis
Other targets of intervention should be taken into account especially in stage 3 to 4 CKD and dialysis patients to reduce the progression of CVD. These targets should include the oxidative stress and inflammation that are well-known features of CKD and kidney failure both with and without replacement therapy [4].
The progressive decline in kidney function (GFR of <15 mL/min/1.73 m2) associated with uremic patients entails accumulation of uremic toxic compounds in the bloodstream and ESRD patients require renal replacement therapy, which is primarily hemodialysis or hemofiltration [62]. These methods require a dialytic filter and a dialysis solution that act to clear toxic molecules from blood and provide concurrent intake of essential solutes. Unfortunately, dialytic procedures cause physical and chemical stress to the vasculature and blood cells: after leaving the vessels to pass toward the membrane in the dialysis circuit, blood loses the protective effect of the endothelium, and both contact with the synthetic surfaces and the change of flow path geometry induce activation of leukocytes and oxidative stress through the release of O2−• and H2O2 [63]. Several types of membranes are available for extracorporeal dialysis, including functionalized membranes with the aim to improve biocompatibility and vascular protection. Dialyzers coated with vitamin E have been shown to provide antioxidant protection to circulating blood cells and lipoproteins [64]. Confirmation of this finding comes from studies in our cohort of patients with kidney failure undergoing dialysis with a vitamin E-coated dialyzer, where we observed a significant reduction of biomarkers related to oxidative stress and inflammation [65]. In particular, p22phox was significantly reduced after 6 months of treatment and declined further after 12 months, along with LDL, plasminogen activator inhibitor-1 (PAI-1), and phosphorylation of ERK 1/2; in contrast, the antioxidant HO-1 was increased [65]. In order to reduce oxidative stress in dialysis patients, a different approach might include supplementation with an oral antioxidant. In this regard, previous studies from our group showed that additional antioxidant supplementation with green tea in patients undergoing bicarbonate dialysis significantly reduced the oxidative imbalance and related cell signaling activity [48]. In particular, green tea led to a reduction in p22phox protein expression and ERK 1/2 phosphorylation after 6 months of treatment and also induced an HO-1 expression together with a reduction in left ventricular mass. In addition, in that study, we found that additional treatment reduced left ventricular mass in patients with LVH and that the reduction was positively correlated with reduction of oxLDL [48].
Hemodiafiltration with online regeneration of ultrafiltrate (HFR) for renal replacement is also effective in reducing oxidative stress. The HFR procedure consists of a double-chambered hemodialysis filter that allows the reinfusion of the ultrafiltrate regenerated through a charcoal-resin cartridge that specifically absorbs proinflammatory cytokines such as IL-6, TNF-α, and CRP. We found that in patients undergoing HFR, compared to patients undergoing standard bicarbonate dialysis, levels of the oxidative stress-related proteins p22phox, PAI-1, and oxLDL were all reduced. In addition, HFR promotes activation of antioxidant defenses such as HO-1 [66].
While in hemodialysis the dialytic filter consists of a synthetic membrane, in peritoneal dialysis the peritoneum itself serves as a natural semipermeable membrane.
Nonetheless, peritoneal dialysis likewise induces increased oxidative stress, as demonstrated by increased AGEs and other prooxidant glucose by-products [67]. In a recent study, we found significantly increased levels of p22phox, ROCK activity (MYPT-1 phosphorylation), and ferritin after 6 months of peritoneal dialysis procedure [68]. These finding call attention to the need for more biocompatible dialysis solutions with different glucose polymers to prevent and/or treat oxidative stress and inflammation. Very preliminary data (not shown) from our laboratory seems to indicate icodextrin reduces oxidative stress in patients undergoing peritoneal dialysis, providing a rationale to follow this path.
Conclusions
The term cardiovascular-renal remodeling describes the complex relationship between the kidney and heart in CKD. It is clear that there is significant interplay between the processes involved in the progression of kidney injury and of CVD that acts to worsen the prognosis of patients. Among these factors, oxidative stress (in terms of excessive ROS, reduced antioxidant defenses, and inflammation) is omnipresent. The few prospective trials and retrospective analyses published to date have provided no conclusive data regarding their treatment, although there is plenty of evidence that these mechanisms are accountable for the onset and progression of CVD in CKD. These pathways should be regarded as important therapeutic targets for the treatment of CKD.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Funding
This study has been supported in part by a grant (DOR 2084023/2020) from the University of Padova to Lorenzo A. Calò.
Authors’ contributions
Conceptualization: VR, LAC
Data curation, Investigation: GB
Resources: LFS, FN
Writing–original draft: VR, GB
Writing–review & editing: LAC, FN
All authors read and approved the final manuscript.