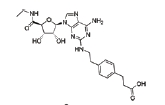
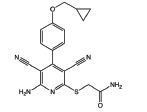
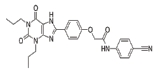
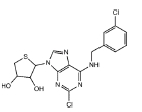
Adenosine receptors as emerging therapeutic targets for diabetic kidney disease
Article information

Abstract
Diabetic kidney disease (DKD) is now a pandemic worldwide, and novel therapeutic options are urgently required. Adenosine, an adenosine triphosphate metabolite, plays a role in kidney homeostasis through interacting with four types of adenosine receptors (ARs): A1AR, A2AAR, A2BAR, and A3AR. Increasing evidence highlights the role of adenosine and ARs in the development and progression of DKD: 1) increased adenosine in the plasma and urine of diabetics with kidney injury, 2) increased expression of each of the ARs in diabetic kidneys, 3) the protective effect of coffee, a commonly ingested nonselective AR antagonist, on DKD, and 4) the protective effect of AR modulators in experimental DKD models. We propose AR modulators as a new therapeutic option to treat DKD. Detailed mechanistic studies on the pharmacology of AR modulators will help us to develop effective first-in-class AR modulators against DKD.
Introduction
Diabetic kidney disease (DKD) is a major diabetic complication and the leading cause of chronic kidney disease (CKD) and end-stage kidney disease worldwide [1]. The mainstay treatment of DKD includes glycemic control, blood pressure control, and blockade of renin-angiotensin-aldosterone system [1]. The current prevalence of DKD implies that these strategies may not fully target the underlying pathogenesis of DKD. Sodium-glucose cotransporter 2 (SGLT2) inhibitor has recently been added as a new therapeutic option against DKD [2]. Nevertheless, we need to develop novel therapeutic strategies targeting DKD through understanding its pathogenesis.
Adenosine is an important endogenous extracellular signaling molecule that is intended to keep or restore homeostasis [3]. Adenosine activates P1 purinergic A1, A2A, A2B, and A3 adenosine receptors (ARs), which are G-protein coupled receptors (GPCR) [4]. All four types of AR are expressed in the kidney, along with metabolically active organs and the immune system [5,6]. Adenosine regulates physiological processes such as glomerular filtration rate (GFR) and homeostasis of water and sodium [7]. Chronically increased adenosine, however, plays a role in various tissue injury [8]. Caffeine, a commonly ingested nonselective AR antagonist, inhibits development of hepatic fibrosis [9] and DKD [10], suggesting AR antagonists as new therapeutic agents against fibrosis including DKD [11]. Increasing evidence highlights the role of adenosine and ARs in the development and progression of DKD [12–23].
Since extracellular adenosine is regulated by membrane transporters as well as conversion from adenosine triphosphate (ATP), the role of conversion of extracellular ATP to adenosine [5] and agents affecting adenosine transport [24] with respect to kidney diseases including DKD have been recently reviewed. In the present review, we provide a background of adenosine and ARs, summarizing the current state of AR modulators in preclinical and clinical evaluation targeting DKD.
Adenosine and adenosine receptors
Adenosine: formation and metabolism
Adenosine is an endogenous purinergic nucleoside. Intracellular adenosine is synthesized either by the dephosphorylation of adenosine monophosphate (AMP) via intracellular 5’-nucleotidase (5’NTD) or by the hydrolysis of S-adenosylhomocysteine (Fig. 1). Extracellular adenosine is produced by dephosphorylation of ATP via a two-step enzymatic reaction sequence. First, CD39 converts ATP or adenosine diphosphate into AMP. In the second step, CD73 converts AMP into adenosine. Adenosine deaminase (ADA) catabolizes adenosine into inosine, and equilibrative nucleoside transporters (ENTs) carry adenosine across the cell membrane in either direction (Fig. 1). Extracellular adenosine is low under normal conditions (20–300 nM) but rises dramatically upon tissue injury, such as hypoxia and inflammation, due to an increased demand for energy supplied by ATP [25]. Increased adenosine up to a certain level provides protection through increased blood flow via vasodilation and through anti-inflammatory/immune-modulatory cascade [26]. However, a chronically increased level of adenosine may play a role in tissue injuries such as hepatic steatosis, asthma, and fibrosis [8]. In fact, persistent and excessive adenosine exposure under ADA deficiency leads to kidney fibrosis [27]. In addition, adenosine uptake is reduced in kidney proximal tubular cells under diabetic stress resulting from reduced ENT, and ENT deficiency leads to kidney fibrosis [19].
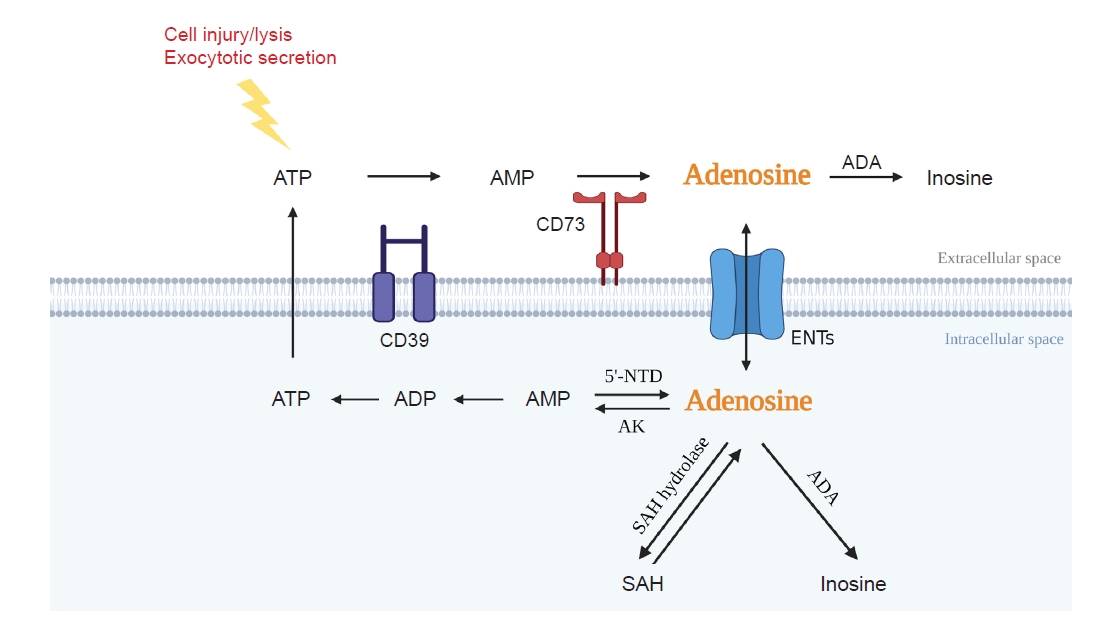
Adenosine: formation and metabolism.
Intracellular adenosine is generated from SAH hydrolase or 5’NTD and is degraded by ADA and AK. Extracellular adenosine is generated by CD73 and converted to inosine by ADA. ENTs allow adenosine flux through the cell membrane depending on gradient concentration.
ADA, adenosine deaminase; AK, adenosine kinase; AMP, adenosine monophosphate; ATP, adenosine triphosphate; ENT, equilibrative nucleoside transporter; SAH, S-adenosylhomocysteine; 5’NTD, 5’-nucleotidase.
Adenosine receptors: classification and signal transduction
Each AR exhibits differences in affinity for adenosine, G protein coupling, and subsequent intracellular signal transduction (Fig. 2).
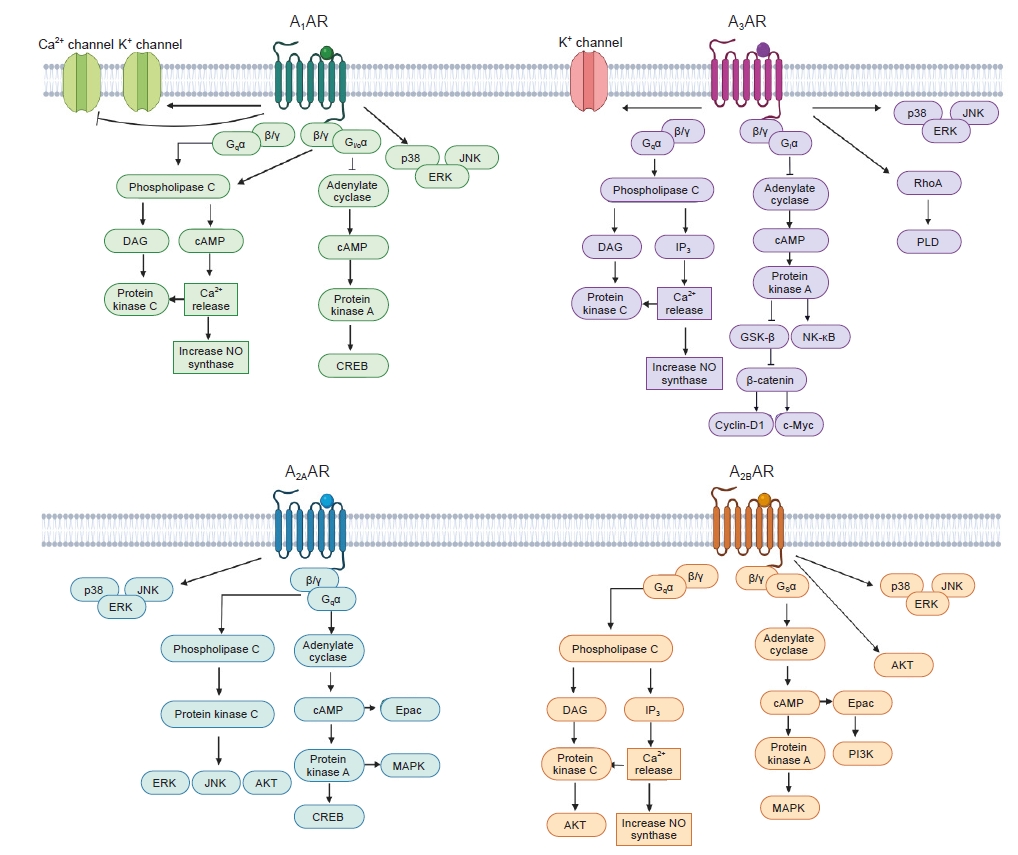
Signaling cascade of each AR.
AR, adenosine receptor; cAMP, cyclic adenosine monophosphate; CREB, cAMP-response element-binding protein; DAG, diacylglycerol; Epac, exchange proteins activated by cAMP; ERK, extracellular signal-regulated protein kinase; GSK-3β, glycogen synthase kinase 3 beta; IP3, inositol trisphosphate; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; NO, nitric oxide; PI3K, phosphatidylinositol 4,5-bisphosphate; PLD, phospholipase D.
Molecular structure of adenosine receptors
All four ARs belong to GPCR, which contains a core domain crossing the plasma membrane seven times (seven transmembrane domains, 7TM), an extracellular N-terminus, and an intracellular C-terminus. The 7TM receptors consist of three intracellular and three extracellular loops with different lengths. Cysteine amino acids forming disulfide bonds in the extracellular loop 2 are important not only in ligand binding but also in receptor stability and function. The N-terminus possesses glycosylation sites, while the C-terminus possesses phosphorylation and palmitoylation sites. These modifications are all important for structural maturation, ligand binding, and downstream signaling [4]. Accordingly, diabetes-induced posttranslational modification, such as glycosylation and palmitoylation, should be considered as a factor that can modify the characteristics of ARs in diabetic kidneys.
All four ARs have been cloned from various species, including rat, mouse, and human. There is a close similarity between species of the same subtype, at least among mammals. However, A3AR shows the largest variability as follows: human vs. rat 73%, human vs. mouse 72%, and rat vs. mouse 91% [4]. Considering rats and mice are the most widely employed animals in preclinical studies, more attention is needed to translate the data from animal studies on A3AR modulators to clinical trials [28].
Classification of adenosine receptors: affinity, G protein coupling, and signal transduction (Fig. 2)
1) A1AR
Adenosine interacts with A1AR with an EC50 in the range of 10 nM to 1 μM. A1AR is coupled to the inhibitory G protein (Gi), which inhibits adenylyl cyclase (AC) activity and subsequent cyclic AMP (cAMP) production [29]. This leads to the inhibition of cAMP-dependent protein kinase A (PKA) activation and subsequent cAMP-responsive element-binding protein (CREB) transcriptional activation by reducing CREB phosphorylation. A1AR also induces phospholipase C (PLC) activation by coupling to the Gq, thus leading to increases in inositol 1,4,5-triphosphate (IP3) and intracellular Ca2+ concentrations, which activate calcium-dependent protein kinases (PKC). In addition, the βγ subunit of Gi/o stimulates PLC. A1AR directly activates K+ channels and inhibits voltage-gated Ca2+ channels [25,29]. The involvement of A1AR and the cascade of mitogen-activated protein kinase (MAPK) family members, including extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), has also been demonstrated in human A1AR-overexpressed CHO cells [30].
2) A2AAR
Adenosine interacts with A2AAR with an EC50 in the range of 10 nM to 1 μM. A2AAR is coupled to the stimulatory G protein (Gs), which stimulates AC and thereby increases cAMP levels with consequent activation of PKA, CREB phosphorylation, exchange protein activated by cAMP (Epac) signaling, and MAPK family [30]. A2AAR stimulates PLC leading to activation of PKC, ERK, JNK, and AKT [31].
3) A2BAR
Adenosine interacts with A2BAR with an EC50 in the range higher than 10 μM. A2BAR is coupled to Gs and Gq [29]. Gs activates AC, causing phosphorylation of PKA and activation of Epac [32]. Gq activates PLC, leading to increase of Ca2+ concentration [29]. A2BAR also activates MAPK and AKT pathways [30].
4) A3AR
Adenosine interacts with A3AR with an EC50 in the range of 10 nM to 1 μM. A3AR is coupled to Gi and Gq [29]. Gi inhibits AC, causing reduced cAMP and PKA leading to activation of glycogen synthase kinase-3β (GSK-3β) and inhibition of beta-catenin [33,34], while Gq stimulates PLC leading to increase of diacylglycerol and intracellular Ca2+ concentrations, cytosolic Ca2+ activates nitric oxide synthase (NOS) or PKC [29]. In addition, A3AR also stimulates RhoA, phospholipase D, ERK, PI3K, and AKT pathways [30].
The role of adenosine and adenosine receptors in the kidney
The kidney is a fundamental organ that maintains the homeostasis by glomerular filtration, reabsorption, and excretion of solutes, ions, and water. The kidney releases diverse hormones that regulate blood pressure, produce red blood cells, and control calcium metabolism [35]. Adenosine, a kidney hormone, participates in such kidney-mediated fundamental physiological functions in homeostasis via regulating kidney blood flow, GFR, tubuloglomerular feedback (TGF), and secretion of renin [7]. However, overproduction of adenosine alters their signaling via the ARs and plays a key role in chronic inflammation, fibrosis, and kidney damage [8,36].
Distribution of adenosine receptors in the kidney
Data from functional studies, immunohistochemistry, and transgenic AR mice show all four ARs are expressed in the kidney [19,36–39]. As summarized in Fig. 3A, all four ARs are expressed in whole kidney homogenates [38], the glomeruli [19,37,39], the proximal tubules [19,40], and the afferent/efferent arterioles [41]. The loop of Henle, distal convoluted tubules, and collecting duct express A1AR/A2AAR [37,38], A1AR/A2BAR [37,38], and A1AR [37], respectively. A recent single-cell transcriptomic analysis study [42] provided a comprehensive atlas of AR expression in the healthy mouse kidney (Fig. 3B).

Distribution of ARs in the kidney.
(A) Summary of each AR expressed in the nephron. (B) Expression of each AR in healthy mouse kidney delineated by single-cell transcriptomic analysis (https://susztaklab.com).
A-IC, alpha intercalated cell; ALOH, ascending loop of Henle; AR, adenosine receptor; B-IC, beta intercalated cell; CD-PC, collecting duct principal cell; CD-Trans, CD transient cell; CNT, connecting tubule; DC 11b+, CD11b+ dendritic cell; DCT, distal convoluted tubule; DLOH, descending loop of Henle; Endo, endothelial; Fib, fibroblast; GEC, glomerular endothelial cells; Granul, granulocyte; LOH, loop of Henle; Macro, macrophage; Neutro, neutrophil; NK, natural killer cell; pDC, plasmacytoid DC; Podo, podocyte; PT, proximal tubule; Tgd, gamma delta T cell; Treg, regulatory T cell.
Physiological role of kidney adenosine receptors
Adenosine directly acts on the coronary arteries to increase blood flow by four times compared to that of at rest [43]. Interestingly, exogenous adenosine induces a marked but transient reduction in the kidney blood flow [44]. Blockade of endogenous adenosine signaling by theophylline-mediated AR antagonism does not alter basal kidney function; however, the exogenous adenosine-mediated reduction of GFR is inhibited with theophylline by 31.4% [45]. All AR subtypes are expressed in the kidney vasculature, yet levels of A1AR and A2BAR are high in the preglomerular microcirculation, whereas A2AAR and A3AR levels are low [46,47]. Adenosine is also involved in the regulation of the TGF of the nephron and renin release from juxtaglomerular cells [48,49].
Vasoconstriction and vasodilation
Pharmacological blockade of the renin-angiotensin system diminishes the kidney vasoconstrictive action of adenosine [45,50], and activation of the renin-angiotensin system augments adenosine-induced vasoconstriction and lowering of GFR [7,50,51]. Selective A1AR antagonists inhibit the adenosine-mediated vasoconstriction, which is absent in A1AR knockout mice [45,52,53]. A1AR-mediated renal vasoconstriction is also supported by the persistent reduction in both kidney blood flow and GFR induced by the infusion of the A1AR agonist [54]. In isolated perfused afferent arterioles, adenosine induces a 30% reduction of vessel diameter in proximal parts of the arteriole [55]. In contrast to A1AR-mediated persistent vasoconstriction in the afferent arterioles, the elevation of kidney adenosine may result in vasorelaxation via A2AR-mediated generation of nitric oxide [53]. A2AAR agonists reduce renal vascular constriction without altering urine volume, sodium excretion, or renin release [56]. A2AAR signaling plays a role in increasing medullary blood flow [57,58]. A3AR agonists do not display vasodilatory influence on isolated afferent arterioles but induce vasodilation in A1AR-induced constricted isolated afferent arterioles [47], suggesting the counterbalancing effects between A1AR and A3AR on afferent arterioles.
Vasoconstriction induced by adenosine or A1AR agonists is blocked by pretreatment with pertussis toxin, indicating that Gi/o protein is involved. The PLC but not the AC signaling pathway plays a role in A1AR-mediated vasoconstriction in the afferent arterioles, since 1) adenylate cyclase inhibitor or PKA inhibitor does not induce vasoconstriction and 2) PLC inhibitor blocks adenosine-induced vasoconstriction in perfused afferent arterioles [59]. Adenosine increases intracellular calcium concentration leading to activation of NOS in afferent arterioles [60]. A1AR-mediated renal vasoconstriction is, in fact, aggravated by NOS inhibitor, suggesting that adenosine activates NOS to counteract A1AR-mediated vasoconstriction [61].
Tubuloglomerular feedback
GFR, defined as the total volume of fluid filtered from the kidney glomeruli in a given period of time, is considered the optimal index for kidney function. TGF is a mechanism that helps to regulate single nephron GFR with the tubular transport activity or capacity. Adenosine signaling triggered by an increased sodium chloride concentration in distal tubules has been suggested to mediate TGF [62].
An intact TGF mechanism requires local adenosine responding to the sodium chloride concentration in the tubular fluid at the macula densa [63]. Sodium levels in urine are sensed by the Na-K-2Cl cotransporter (NKCC2) in the macula densa. Activated NKCC2 leads to synthesis of adenosine and elevated adenosine levels in glomerular capillaries. Adenosine induces the contraction of afferent arterioles and dilation of efferent arterioles through activation of adenosine A1AR and A2AR, respectively, as described above; subsequently, single nephron GFR and the filtration pressure are suppressed. Regarding this, A1AR knockout mice lack the TGF mechanism [64,65]. A2AAR in efferent arterioles induces vasodilation and leads to reducing GFR [66]. Administration of an A2AAR antagonist increases GFR without a change in renal blood flow, suggesting a tonic influence of endogenous adenosine on efferent arterioles [14].
Secretion of renin
Adenosine inhibits secretion of renin in the kidney [7,67]. A1AR antagonists increase plasma renin level, indicating a tonic inhibition of renin secretion by A1AR activation [68]. Consistently, A1AR knockout mice show increased renin expression and content in the kidney [69]. A2AAR activation, however, increases renin secretion [70].
The role of adenosine and adenosine receptors in the diabetic kidney disease
Plasma levels of adenosine and its catabolic product, inosine, are increased in patients with DKD [21,22]. Interestingly, plasma adenosine is not different between healthy and diabetic patients without kidney disease [21]. Our urinary analysis of diabetic patients showed proportional increases in urinary adenosine excretion according to the stage of DKD (Fig. 4A). In addition, urinary excretion of adenosine shows positive correlation with proteinuria or microalbuminuria and negative correlation with creatinine clearance (data not shown). Levels of adenosine in kidney tissue and urine are increased in streptozotocin (STZ)-induced diabetic mice and rats [16,71]. The isolated glomeruli from diabetic rats contains six-fold higher adenosine compared to controls [72]. Adenosine in renal venous plasma but not in renal arterial plasma is significantly increased in diabetic rats compared to controls [23], further suggesting increased synthesis and release of adenosine in diabetic kidneys. Additionally, kidney expression of CD73 transcription and protein are increased in STZ-induced diabetic mice and rats [16,71]. The expression of kidney adenosine kinase (AK) is decreased in diabetic rats, whereas administration of insulin to diabetic rats restores the kidney AK expression [73,74]. The administration of ADA in diabetic rats decreases the urinary excretion of proinflammatory cytokines and increases the anti-inflammatory cytokine [20], suggesting a pathogenic role of increased adenosine in DKD. The ENT, which modulates adenosine uptake, is reduced in diabetic kidneys, contributing to kidney fibrosis [19].
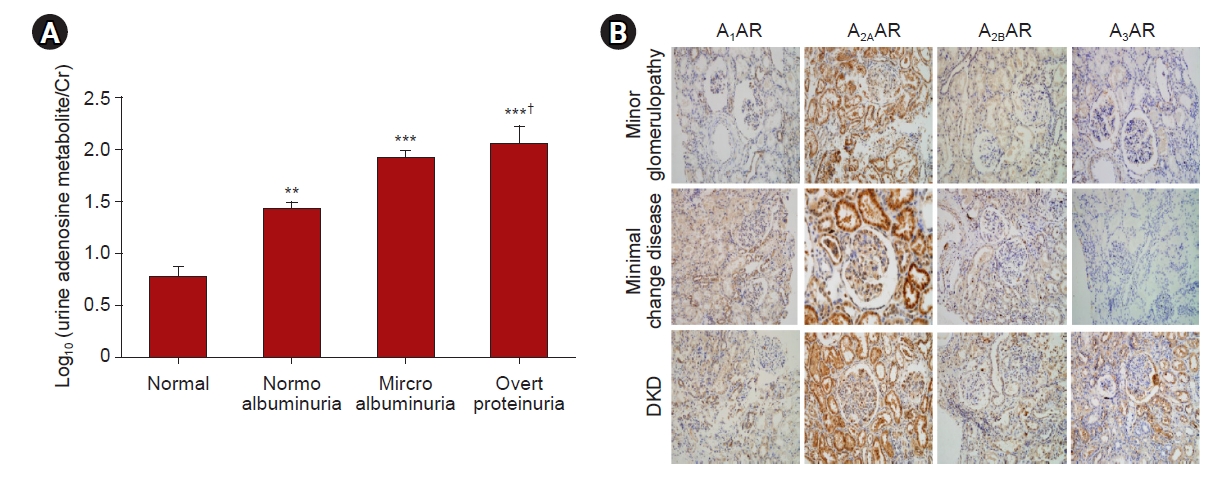
Urinary excretion of adenosine and renal expression of ARs in diabetic patients.
(A) Urinary excretion of adenosine from normal (n = 10), normoalbuminuria (n = 10), microalbuminuria (n = 10), and overt proteinuria (n = 10) were measured. (B) Immunostaining of ARs was performed from minor glomerulopathy, minimal change disease, and DKD. Original magnification ×400. The data are presented as the mean ± standard error of mean.
AR, adenosine receptor; Cr, creatinine; DKD, diabetic kidney disease.
**p < 0.01, ***p < 0.01 vs. control; †p < 0.05 vs. normoalbuminuria.
In contrast to the above studies, administration of adenosine, inhibition of cellular uptake of adenosine, and blockade of AK have all been shown to reduce kidney injury in diabetes [75–77]. In addition, CD73 knockout diabetic mice exhibited more severe kidney injury than wild-type diabetic mice [16]. The exact reason for these seemingly contradictory findings is not clear but may be due to experimental conditions such as duration of diabetes and severity of DKD.
We confirmed that expression of each AR is significantly increased in both STZ-induced type-1 diabetes (T1D) and db/db type-2 diabetes (T2D) mouse kidney (Fig. 5A, B). In addition, analysis of the Nephroseq database of human diabetic kidney biopsy samples showed increased AR expression (Fig. 5C). Our immunohistochemical analysis shows that A2AAR expression is increased in minor glomerulopathy, minimal change disease, and DKD. Interestingly, A3AR expression is significantly increased only in DKD, and not in minor glomerulopathy or minimal change disease (Fig. 4B).

The expression of ARs in diabetic kidney.
(A, B) Messenger RNA (mRNA) expressions of ARs were obtained from kidney homogenate of STZ-induced diabetic mice (12 weeks after STZ injury) and 20-week-old db/db mice. (C) Microarray analyses of human kidney biopsy data were provided in the Nephroseq database (nephroseq.org). The data are presented as the mean ± standard error of mean.
AR, adenosine receptor; ERCB, European Renal cDNA Bank; STZ, streptozotocin.
*p < 0.05 vs. control, db/m, normal.
The involvement of adenosine and ARs in DKD has been also studied using genetically engineered models. Lack of A1AR in diabetic mice aggravates both hyperfiltration and glomerular injuries [78]. Both A2AAR or A2BAR knockout diabetic mice exhibited more severe kidney injury, as indicated by increased albumin excretion compared to wild-type diabetic mice [13,16]. The severity of DKD in A3AR knockout mice has not been reported, while A3AR knockout mice are protected against ischemia- or myoglobinuria-induced AKI [79]. Considering the ubiquitous distribution and diverse effects of each AR, future studies with spatiotemporal regulation of AR during the progression of DKD are necessary to understand their exact roles in diabetic kidneys.
The role of NKCC2/adenosine/AR in the renoprotective effect of SGLT2 inhibitors was particularly interesting. SGLT2 inhibitors have recently been shown to prevent the progression of DKD [80,81]. SGLT2 inhibitors reduce proximal reabsorption of sodium chloride and increase sodium chloride concentrations at the macula densa, facilitating NKCC2 transporter which subsequently leads to production of adenosine, activates the TGF mechanism, and reduces hyperfiltration (summarized in section of Tubuloglomerular feedback) associated with DKD [82]. It is speculated that upregulated SGLT2, even in the presence of hyperfiltration in diabetic kidney, will reduce sodium chloride delivered to macular densa, inactivate NKCC2, decrease the adenosine/AR axis in TGF, and maintain hyperfiltration. This speculation is supported by the fact that SGLT2 inhibition in T1D patients increases urinary adenosine excretion [83]. Restoration of TGF as a result of increased extracellular adenosine (due to inhibition of ENT) also has been suggested as a renoprotective mechanism of dipyridamole in STZ-induced diabetic rats [84].
Therapeutic applications of adenosine receptor modulators against diabetic kidney disease
The consumption of more than two cups per day of coffee, which contains the nonselective adenosine antagonist caffeine, for 2 weeks improved eGFR in DKD patients [10], and there is an inverse relationship between caffeine consumption and mortality in CKD patients [85]. In addition, a nonselective AR antagonist, 8-(p-sulfophenyl) theophylline, effectively decreases kidney fibrosis and improves kidney function [86]. These data suggest that well-designed AR antagonists may become novel protective agents against DKD.
With respect to specific AR subtype, A3AR antagonists consistently prevent experimental DKD [18–20] as summarized in Table 1 and Fig. 6. LJ2698 and MRS1220, the A3AR antagonists, attenuate fibrosis, oxidative stress, and inflammation in db/db mice [18] and STZ diabetic rats [19,20], respectively. Our previous study showed that LJ1888, a prototype compound of LJ2698, prevents fibrosis in obstructed kidney as well [87]. Inhibition of transforming growth factor-β1 (TGF-β1)–induced extracellular matrix (ECM) upregulation via A3AR-specific small interfering RNA in renal proximal epithelial cells [87] confirmed the fibrotic effects of A3AR in the kidney. We have shown that LJ1888 reduced obstruction- or TGF-β1-induced upregulation of lysyl oxidase, which induces cross-linking of ECM, suggesting that A3AR antagonists may also regulate ECM accumulation via posttranslational regulation [87]. Yet, more studies are needed to confirm the therapeutic effect of A3AR antagonists against DKD, because decreased proinflammatory cytokines in response to A3AR agonists and increased proinflammatory cytokines in A3AR knockout mice under sepsis-induced kidney injury have been reported [88].

Structure and efficacy of AR modulators against DKD
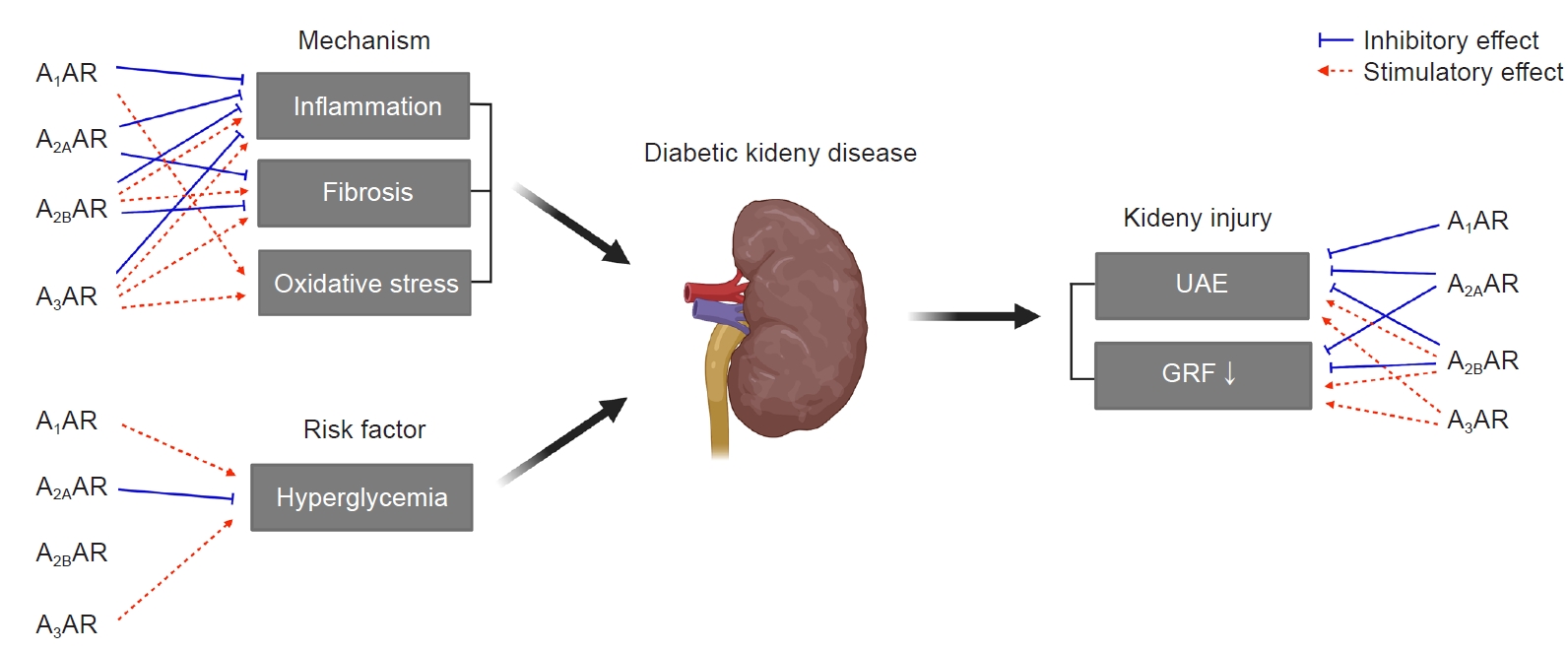
The role of ARs in diabetic kidney disease.
Each AR has either stimulatory (red dashed arrows) or inhibitory (blue solid inhibitory lines) effects on inflammation, fibrosis, oxidative stress, hyperglycemia, UAE, and GFR in diabetic kidneys.
AR, adenosine receptor; GFR, glomerular filtration rate; UAE, urinary albumin excretion.
Inflammation plays a key role in the development of DKD [1], and A2AAR acts as a strong anti-inflammatory effector responding to extracellular adenosine [89]. In agreement with this, the A2AAR agonists ATL146 or CGS21680 attenuate kidney injury including inflammation as well as fibrosis in STZ-induced diabetic rats [13–15]. A2AAR activation also reduces cytokine and chemokine expression in the kidney tubular epithelial cells under hypoxia and inhibits the macrophage infiltration after ischemia-reperfusion [90,91]. A2AAR activation, in fact, attenuates kidney fibrosis and suppresses the epithelial-mesenchymal transition in various kidney fibrosis models [92,93].
Since A3AR antagonists or A2AAR agonists protect against DKD, it is reasonable to develop AR modulators with dual action as an A3AR antagonist and an A2AAR agonist in order to have better protective effects against DKD. We showed for the first time that LJ4459, a newly synthesized dual-acting ligand, prevents the progression of tubulointerstitial fibrosis in obstructed kidneys [94]. It will be interesting to determine whether this dual-acting ligand has better therapeutic efficacy against DKD than an A3AR antagonist or an A2AAR agonist.
Opposing pro- and anti-therapeutic roles of A2BAR in DKD have been reported. The A2BAR agonist Bay60-6583 attenuates diabetic kidney fibrosis [16]. On the contrary, 1) Bay60-6583 promotes profibrotic activation of kidney fibroblasts [95] and 2) MRS1754, the A2BAR antagonist, attenuates profibrotic effects along with reduced vascular endothelial growth factor expression in diabetic rats and mice [12,17]. An anti-inflammatory role of A1AR in ischemia/reperfusion kidney injury has been reported [96–98], but the role of A1AR modulators in DKD remains to be studied.
Oxidative stress plays a key role in the development and progression of DKD [99]. Diabetic stress including high glucose and lipids increases reactive oxygen species (ROS), and mitochondrial dysfunction and nicotinamide adenine dinucleotide phosphate oxidase (NOX) play key, if not all, roles in increased ROS under diabetic conditions [100]. A3AR antagonists reduce adriamycin-induced NOS upregulation in the kidney [101], and A1AR or A3AR knockout reduces NOX-derived oxidative stress in mice [102,103].
The involvement of adenosine on metabolism should be considered, since hyperglycemia is a key player in DKD [1]. The adenosine system has been suggested to play a role in glucose homeostasis, contributing to the pathophysiology of T1D and T2D [3]. Caffeine reduces plasma glucose and increases pancreatic insulin in STZ diabetic rats [104]. Pharmacological inhibition of A1AR or knockout of A1AR in mice enhances basal insulin secretion [105,106]. On the other hand, A2AAR activation increases insulin secretion and improves beta-cell function by inhibiting the inflammatory response [107,108]. The absence of A3AR in mice improved a metabolic phenotype including reduced glucose clearance and increased insulin compared to wild-type mice [103] (Fig. 6). Although further mechanistic studies are required, activation of A2AAR and inhibition of A1AR or A3AR improves insulin resistance and hyperglycemia, which may play a role in preventing DKD.
While various AR agonists or antagonists are under clinical trials, according to clinicaltrials.gov, targeting inflammatory, cancer, and cardiovascular diseases, none of the AR modulators are U.S. Food and Drug Administration-approved or under clinical trials against DKD at present.
Conclusion
The severity of DKD and limited therapeutic modality underscore the urgency to develop new therapeutic strategies based on pathogenesis of DKD. In this review, we summarized the (patho)physiological role of adenosine and ARs in diabetic kidneys and updated AR modulators showing a protective effect in experimental DKD. There has been no AR modulator in clinical trials against DKD. Considering 1) the protective effect of caffeine on kidney injury in diabetic patients, 2) increased plasma and urinary excretion of adenosine under diabetic stress, 3) upregulation of expression of each AR in diabetic kidneys, and 4) the protective effect of AR modulators in experimental DKD models, we propose AR modulators as a new therapeutic option to treat DKD. Detailed mechanistic studies on the pharmacology of AR modulators, including the role of diabetes-induced posttranslational modification such as glycosylation and palmitoylation, in AR function will help us to develop effective first-in-class AR modulator agents for DKD.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Funding
The preparation of this manuscript was supported by a National Research Foundation grant (No. 2020R1A6A3A13076183), Republic of Korea, and by Ewha Womans University (No. 1-2021-1301-001-1).
Authors’ contributions
Conceptualization, Funding acquisition: ESP, HH
Supervision: HH
Writing–original draft: ESP
Writing–review & editing: All authors
All authors read and approved the final manuscript.