Long non-coding RNA MEG3 silencing weakens high glucose-induced mesangial cell injury by decreasing LIN28B expression by sponging and sequestering miR-23c
Article information

Abstract
Background
Diabetic nephropathy (DN) is a common kidney disease in diabetic patients. Long non-coding RNA maternally expressed gene 3 (MEG3) and microRNA (miR)-23c are reported to be implicated in DN development. Nevertheless, it is unclear that the molecular mechanism between MEG3 and miR-23c in DN remains unclear.
Methods
Human mesangial cells (HMCs) were treated with high glucose (HG) to simulate the DN status in vitro. Expression of MEG3 and miR-23c was measured. Effects of MEG3 silencing on HG-stimulated HMC injury were determined. The relationship between MEG3 and miR-23c was verified by the dual-luciferase reporter and RNA immunoprecipitation assays.
Results
MEG3 was overexpressed in serums from DN patients and HG-stimulated HMCs. MEG3 knockdown weakened HG-stimulated HMC proliferation, extracellular matrix (ECM) accumulation, and inflammation. MEG3 regulated lin-28 homolog B (LIN28B) expression through adsorbing miR-23c. MiR-23c inhibitor reversed MEG3 knockdown-mediated effects on HG-stimulated HMC proliferation, ECM accumulation, and inflammation. LIN28B overexpression overturned miR-23c mimic-mediated effects on HG-stimulated HMC proliferation, ECM accumulation, and inflammation.
Conclusion
MEG3 regulated HMC injury via regulation of the miR-23c/LIN28B axis in DN, which can help us better understand the mechanism of DN mediated by MEG3.
Introduction
Diabetic nephropathy (DN), a common diabetic complication, is the main cause of end-stage renal disease [1]. DN is related to the structural changes of the kidney, which is manifested by the thickening of the glomerular basement membrane and the accumulation of extracellular matrix (ECM) [2]. Currently, hyperglycemia is recognized as the main driving force for the development of DN. Studies have uncovered that hyperglycemia can cause inflammation, which promotes the development of DN [3,4]. Also, high glucose (HG) induces proliferation and ECM accumulation in mesangial cells [5,6]. As reported, epithelial-to-mesenchymal transition (EMT) accelerates the accumulation of ECM components [7,8].
Experiments and clinical studies on mesangial cells have confirmed that the phenotypic transformation of mesangial cells exists in DN, characterized by mesangial hypertrophy and massive secretion of ECM [9,10]. Phenotypic transformation of the mesangium may be the cytological basis for the gradual accumulation of ECM in the glomerulus, eventually resulting in glomerulosclerosis [11,12]. Therefore, exploring the pathogenesis of DN using human mesangial cells (HMCs) is of great significance for understanding the pathogenesis of the disease.
Long non-coding RNAs (lncRNAs) are a type of non-coding transcripts that exert vital roles in the development of many diseases, including DN [13,14]. For instance, lncRNA NR_033515 facilitated DN progression via accelerating proliferation, EMT, and fibrogenesis of mesangial cells [15]. Also, lncRNA plasmacytoma variant translocation 1 could contribute to podocyte damage and apoptosis in DN [16]. LncRNA maternally expressed gene 3 (MEG3) is located at chromosome 14q32.3 [17]. It has been reported that MEG3 plays a vital role in some diseases. Mounting studies revealed the tumor-inhibiting role of MEG3 in diverse cancers [18]. MEG3 could protect endothelial function by modulating the DNA damage response [19]. Also, MEG3 played an inhibitory role by inactivating the AKT/mTOR pathway in rheumatoid arthritis [20]. In DN, MEG3 could alleviate proliferation, fibrosis, and inflammation of mesangial cells [21,22]. However, the regulatory mechanism of MEG3 on HG-induced mesangial cell damage has not been fully elucidated.
According to the competing endogenous RNA (ceRNA) hypothesis, lncRNAs act as microRNA (miR) sponges and can bind miRs competitively to indirectly regulate messenger RNAs (mRNAs) using shared miR response elements. MiRs can modulate gene expression by inducing translational repression or degradation [23]. MiRs are implicated in almost all pathological and physiological processes, including cell differentiation, immune inflammation, apoptosis, and proliferation [24]. Recent studies have revealed that miRs have great potential as therapeutic targets and diagnostic markers in many diseases, including DN [24]. The starBase predicted that many miRNAs may interact with MEG3. By reviewing previously reported literature, we searched for five miRNAs (miR-145-5p, miR-143-3p, miR-181a-5p, miR-485-5p, and miR-23c) that are low expressed and inhibit mesangial cell proliferation in DN mesangial cells. The miR-23c with the largest decrease in MEG3 overexpression in cells had been selected for further analysis (Supplementary Fig. 1, available online). MiR-23c plays a tumor-repressive role in endometrial cancer [25] and hepatocellular cancer [26]. In DN, high miR-23c expression inhibits pyroptosis of renal tubular epithelial [27] and decreases proliferation, fibrosis, and EMT of mouse mesangial cells [28]. However, the mechanism of miR-23c dysregulation in mesangial cells under HG stimulation is unclear.
Herein, the relationship between MEG3 and miR-23c in HG-induced mesangial cell damage was explored. Our results uncovered that MEG3 inhibition reduced HG-induced proliferation, ECM accumulation, and inflammation in HMCs via upregulating lin-28 homolog B (LIN28B) through adsorbing miR-23c.
Methods
Serum samples
Peripheral blood (5 mL) was obtained from 25 healthy controls (normal) and 25 type 2 diabetes with DN. Twenty-five kidney tissues were collected from type 2 diabetes with DN through ultrasound-guided kidney biopsy. Twenty-five normal kidney samples were obtained from patients who underwent renal tumor resection. All recruited subjects were diagnosed or underwent basic examination at the People’s Hospital of Zhengzhou University. The permission for this research was approved by the Ethics Committee of People’s Hospital of Zhengzhou University (No. 20210628), and all participants signed informed consent. The clinical and biochemical characteristics are exhibited in Table 1.

Clinical and biochemical characteristics of DN patients and healthy controls
Cell culture and treatment
HMCs (BNCC341276; BNCC) were cultured in Dulbecco’s modified eagle medium (DMEM; Sigma-Aldrich) supplemented with glucose (5 mM, #G8270; Sigma-Aldrich), fetal bovine serum (FBS; #10100147, 10%; Life Technologies [LT]), and strep/penicillin (#P0781, 1%; Sigma-Aldrich). For HG stimulation, 30 mM of glucose was used, with 5 mM of glucose serving as a control.
Isolation of human primary mesangial cells
The separated cortical tissue on the superficial surface of the kidney is finely cut into extremely fine pieces and lightly ground with a tissue grinder. The cell suspension was filtered on a 200 mesh stainless steel screen and washed three times with D-Hank’s solution containing penicillin-streptomycin double antibody to remove blood cells as much as possible. The tissue on the sieve was digested using a DMEM medium containing 0.15% type IV collagenase (Sigma-Aldrich). After removing the supernatant, add an appropriate amount of DMEM medium and centrifuge again to obtain the cell precipitation. The cells were cultured in DMEM supplemented with 20% FBS.
Cell transfection
Oligonucleotides or vectors were transfected into HMCs using Lipofectamine 3000 reagent (#L3000001; Invitrogen). Small interfering (si) RNA against MEG3 (si-MEG3: GGUUGUUGUGAGAAUUAAAUG) and its negative control (NC) (si-NC: 5’-UUCUCCGAACGUGUCACGUTT-3’), miR-23c inhibitor and mimic (anti-miR-23c: 5’-GGGUAAUCACUGGCAAUGUGAU-3’ and miR-23c: 5’-AUCACAUUGCCAGUGAUUACCC-3’), anti-miR-NC (5’-CGGUUCGCTAGCGCCGACUCG-3’), and miR-NC (5’-CGAUCGCAUCAGCAUCGAUUGC-3’) were purchased from RiboBio. The full-length complementary DNA (cDNA) of MEG3 (NR_002766) or LIN28B (NM_001004317) was digested with KpnI-XhoI or BamHI-NotI and then ligated into the pcDNA3.1(+) vector (Life Technologies) to construct the MEG3 and LIN28B overexpression plasmid.
Quantitative real-time polymerase chain reaction
Plasma/Serum RNA Purification Mini Kit (#RNB700; Sigma-Aldrich) and RNeasy Mini Kit (#74104; QIAGEN) were used to isolate total RNA from serum samples or HMCs. The M-MLV First Strand Kit (#28025013; Life Technologies) was for cDNA generation. Quantitative PCR was conducted with the SYBR Green PCR Master Mix (#1725270; Bio-Rad). The 2–ΔΔCt method was applied to figure the expression of MEG3, LIN28B, and miR-23c in serum samples or HMCs with β-actin or U6 as a housekeeping gene, and the primers were added to Table 2.

Primer sequences for quantitative real-time polymerase chain reaction
Cell proliferation analysis
The proliferation of HMCs was analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and 5-ethynyl-2’-deoxyuridine (EdU) assays. For MTT assays, HMCs (5 × 103 cells/well) were cultured for 24 hours, 48 hours, or 72 hours. Then, the MTT solution (#CT01-5, 100 μL, 0.5 mg/mL; Sigma-Aldrich) was supplemented into each well. Dimethylsulfoxide (#D8418, 150 μL; Sigma-Aldrich) was added to each well to dissolve the crystals after 4 hours of incubation. Next, the absorbance at 570 nm was analyzed by Microplate Reader (Bio-Rad). The EdU assay was performed using the Cell-Light EdU Kit (#C10310-1; RiboBio) based on the manufacturer’s instructions. Nuclear staining was performed with DAPI (#MBD0015; Sigma-Aldrich), and the cells were observed under a fluorescence microscope (Olympus).
Cell cycle progression analysis
In brief, HMCs were harvested and fixed with ethanol (70%) at 4 °C overnight, followed by incubation with RNase A containing propidium iodide (40%, #81845; Sigma-Aldrich). Cell distribution was assessed with FACScan flow cytometry (BD Biosciences) and FACS Diva Software (BD Biosciences).
Western blotting
HMCs were lysed in radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail (Roche). Total protein of 40 μg was isolated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride (PVDF) membranes (Roche). After blocking with tris buffered saline tween buffer with 5% skim milk, the PVDF membranes were incubated with primary antibodies. Thereafter, the PVDF membranes were incubated with second antibodies. The primary antibodies were exhibited as follows: anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab9484), anti-LIN28B (ab115698), anti-N-cadherin (sc-8424), anti-E-cadherin (sc-8426), anti-collagen IV (sc-59814), and anti-fibronectin (sc-18825). Protein bands were visualized with an ImmunoStar LD (Wako Pure Chemical). The primary antibodies for LIN28B and GAPDH were bought from Abcam, and other antibodies were purchased from Santa Cruz Biotechnology. All original protein bands of western blotting were exhibited in Supplementary Fig. 2 (available online).
Enzyme-linked immunosorbent assay
The levels of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) were detected with the enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems). Briefly, the supernatants of HMCs were collected after culture for 48 hours, followed by the analysis of the absorbance with the Microplate Reader (Bio-Rad).
Dual-luciferase reporter assay
The binding sites of MEG3 or LIN28B in miR-23c were predicted with the starBase database. The luciferase reporters containing wild type (WT)-MEG3, mutant (MUT)-MEG3, LIN28B 3’ untranslated regions (UTR)-WT, or LIN28B 3’ UTR-MUT were synthesized by GenePharma. The luciferase reporter vectors were cotransfected into HMCs together with miR-23c or miR-NC when the cell reached 80% confluence. After 48 hours of transfection, the luciferase activities of Firefly and Renilla were measured with the luciferase reporter assay kit (Promega). The ratio of Firefly luciferase activity to Renilla luciferase activity was the relative luciferase activity.
RNA immunoprecipitation assay
RNA immunoprecipitation (RIP) analysis was executed with the Magna RIP Kit (Millipore). Briefly, about 2 × 107 HMCs were lysed in complete RIP lysis buffer containing protease/RNase inhibitors (Roche) and then centrifuged at 16,000 ×g at 4 °C for 5 minutes. Subsequently, cell extracts were incubated with magnetic beads conjugated with anti-immunoglobulin G (ab172730; Abcam) or anti-Ago2 (ab186733; Abcam). Immunoprecipitated RNAs were isolated by TRIzol (Life Technologies) after the sample was treated with protease K at 55 °C for 30 minutes. The coprecipitated RNAs were purified and then analyzed by quantitative real-time polymerase chain reaction (qRT-PCR).
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was implemented by IBM SPSS version 20.0 (IBM Corp.) with the unpaired Student t test and one-way analysis of variance. Data were exhibited as mean ± standard deviation. A p-value of <0.05 was considered statistically significant.
Results
The negative correlation between MEG3 and miR-23c in diabetic nephropathy
Previous studies have revealed that MEG3 and miR-23c exert opposite roles in DN [22,27]. We detected the expression of MEG3 and miR-23c in serums of DN patients (n = 25) and healthy controls (n = 25) to investigate the relationship between MEG3 and miR-23c. qRT-PCR exhibited higher MEG3 levels in serums derived from DN patients than healthy controls, while miR-23c expression had an adverse result (Fig. 1A, B). Pearson correlation analysis indicated a negative correlation between MEG3 and miR-23c expression in serums derived from DN patients (Fig. 1C). Additionally, we detected MEG3 and miR-23c levels in 25 DN kidney tissues and 25 normal kidney tissues. The results exhibited that MEG3 was observably upregulated in DN kidney tissues than in normal kidney tissues, while miR-23c had an opposing result (Supplementary Fig. 3, available online). MEG3 expression in patients with normoalbuminuria, diabetic microalbuminuria, and macroalbuminuria was elevated successively. However, miR-23c expression had an opposite result (Supplementary Fig. 4A, B; available online). In addition, DN patients with high MEG3 expression had higher fasting blood glucose levels, while DN patients with high miR-23c expression had lower fasting blood glucose levels (Supplementary Fig. 4C, D; available online). Subsequently, we examined MEG3 and miR-23c expression in HG-stimulated HMCs. Likewise, MEG3 expression was elevated while miR-23c expression was decreased in HG-stimulated HMCs relative to the control group (Fig. 1D, E). Taken together, the negative correlation between MEG3 and miR-23c in DN might be related to the progression of DN.
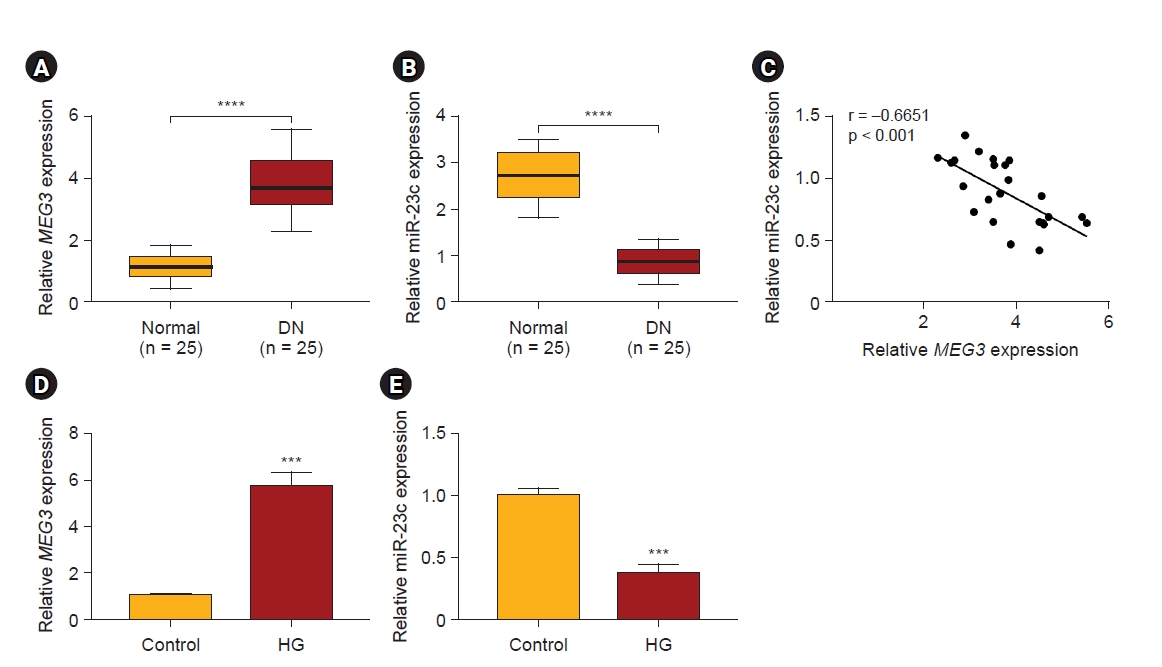
Expression patterns of MEG3 and miR-23c in DN and HG-stimulated HMCs.
(A, B) Expression levels of MEG3 and miR-23c in serums derived from DN patients (n = 25) and healthy controls (n = 25). (C) The correlation between MEG3 and miR-23c in the serum of DN patients (Pearson correlation analysis). (D, E) Expression levels of MEG3 and miR-23c in HG-stimulated HMCs and control group.
DN, diabetic nephropathy; HMC, human mesangial cell; HG, high glucose; MEG3, maternally expressed gene 3; miR, microRNA.
***p < 0.001 and ****p < 0.0001.
MEG3 inhibition reduced high glucose-stimulated human mesangial cell proliferation, extracellular matrix accumulation, and inflammation
Next, we explored the biological role of MEG3 in DN. qRT-PCR exhibited that transfection of si-MEG3 reversed the elevation of MEG3 in HMCs induced by HG treatment (Fig. 2A). MTT and EdU assays showed that MEG3 inhibition weakened HG stimulation-induced HMC proliferation (Fig. 2B, C). Moreover, MEG3 silencing caused HG-induced HMCs to be arrested in G0/G1 phase, suggesting that MEG3 silencing weakened HG-induced cell cycle progression in HMCs (Fig. 2D). Western blotting presented that MEG3 silencing weakened upregulation of fibronectin and collagen IV protein levels in HG-stimulated HMCs (Fig. 2E). Moreover, MEG3 inhibition reversed the downregulation of E-cadherin and the upregulation of N-cadherin in HMCs induced by HG treatment (Fig. 2F). Subsequently, ELISA exhibited that MEG3 downregulation could decrease the release of IL-6 and TNF-α in HMCs mediated by HG (Fig. 2G). Additionally, we surveyed the biological function of MEG3 in mesangial cells isolated from DN kidney tissues. The results exhibited that MEG3 knockdown reduced proliferation and ECM accumulation of mesangial cells in vitro (Supplementary Fig. 5, available online). Together, these findings indicated that MEG3 silencing decreased HMC injury induced by HG treatment.

Effects of MEG3 downregulation on HG-induced HMC injury.
(A–F) HMCs were transfected with si-MEG3 or si-NC and then treated with HG. (A) qRT-PCR analysis of MEG3 expression in HMCs. (B–D) MTT, EdU, and flow cytometry assays were performed to evaluate the proliferation and cell cycle progression of HMCs. (E, F) Western blotting analysis of fibronectin, collagen IV, E-cadherin, and N-cadherin protein levels in HMCs. (G) Levels of IL-6 and TNF-α in HMCs.
EdU, 5-ethynyl-2'-deoxyuridine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HMC, human mesangial cell; HG, high glucose; IL, interleukin; MEG3, maternally expressed gene 3; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NC, negative control; NS, not significant; OD, optical density; qRT-PCR, quantitative reverse transcription polymerase chain reaction; si, small interfering; TNF, tumor necrosis factor.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
MEG3 was verified as a miR-23c sponge
Based on the opposite expression levels of MEG3 and miR-23c in DN, we speculated that MEG3 might be a sponge of miR-23c. Online bioinformatics prediction (starBase) exhibited that MEG3 and miR-23c had complementary binding sites (Fig. 3A). And miR-23c overexpression could reduce the luciferase activity of the luciferase reporter with WT-MEG3 (Fig. 3B). RIP assay manifested that miR-23c and MEG3 were enriched in the anti-Ago2 group (Fig. 3C). After MEG3 transfection, MEG3 expression was intensified in HG-stimulated HMCs (Fig. 3D). Furthermore, MEG3 inhibition weakened the decrease in miR-23c expression in HG-stimulated HMCs, but MEG3 overexpression had an opposing effect (Fig. 3E). Collectively, MEG3 acted as a miR-23c sponge.
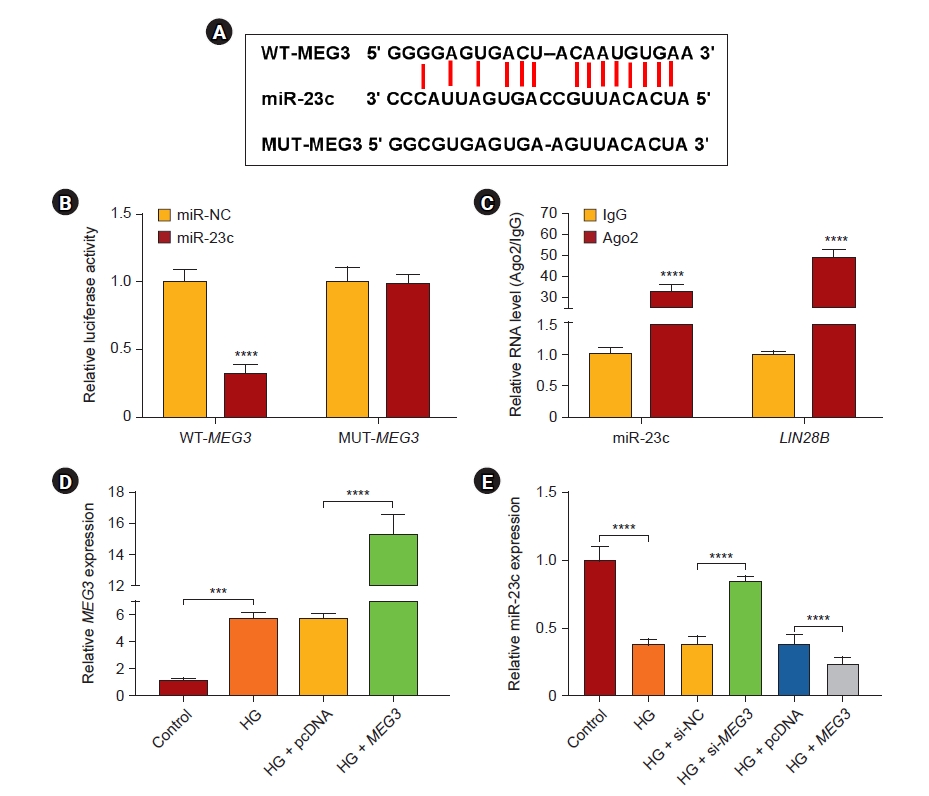
MEG3 was a sponge for miR-23c in HMCs.
(A) The diagram showed the complementary binding sites of miR-23c and MEG3. (B) Dual-luciferase reporter assay was performed to verify the binding sites between miR-23c and MEG3. (C) Levels of miR-23c and MEG3 in RNA-induced silencing complexes were analyzed. (D) Expression of MEG3 in HMCs with pcDNA or MEG3 under HG treatment. (E) Expression of miR-23c in HMCs with si-NC, si-MEG3, pcDNA, or MEG3 under HG treatment.
HG, high glucose; HMC, human mesangial cell; IgG, immunoglobulin G; MEG3, maternally expressed gene 3; miR, microRNA; MUT, mutant; NC, negative control; pcDNA, plasmid DNA; si, small interfering; WT, wild type.
***p < 0.001 and ****p < 0.0001.
MiR-23c inhibitor abolished MEG3 downregulation-mediated effects on high glucose-induced human mesangial cell injury
Subsequently, we further surveyed whether miR-23c was implicated in DN progression regulated by MEG3. qRT-PCR showed that introduction of anti-miR-23c alleviated MEG3 knockdown-mediated downregulation of miR-23c in HMCs induced by HG (Fig. 4A). Moreover, MEG3 silencing resulted in the impaired ability of cell proliferation and the arrest of cell cycle progression in the G0/G1 phase of HG-induced HMCs, but these effects mediated by MEG3 inhibition were lightened after miR-23c silencing (Fig. 4B–E). Also, miR-23c downregulation overturned MEG3 silencing-mediated inhibition on ECM accumulation, EMT, and inflammation of HMCs under HG treatment (Fig. 4F–H). Therefore, these findings revealed that MEG3 could modulate HG-induced HMC injury via sponging miR-23c.
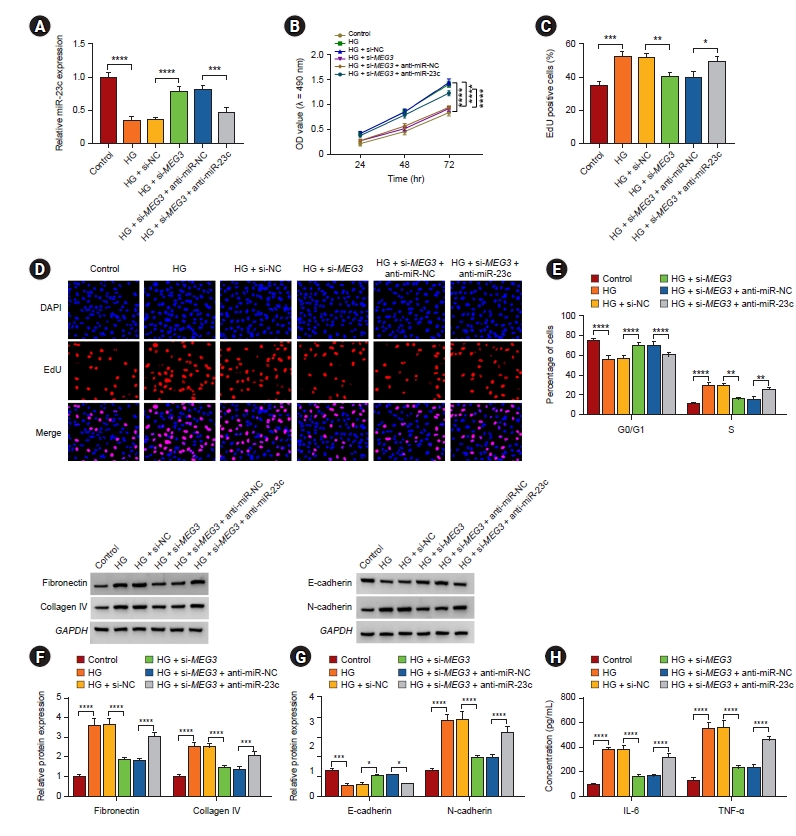
MEG3 accelerated HG-induced HMC injury via sponging miR-23c.
(A–H) After si-NC, si-MEG3, si-MEG3 + anti-miR-NC, or si-MEG3 + anti-miR-23c transfection, HMCs were treated with HG. (A) qRT-PCR analysis of miR-23c expression in HMCs. (B–E) The proliferation and cell cycle progression of HMCs were determined (D, original magnification of ×200). (F, G) Protein levels of fibronectin, collagen IV, E-cadherin, and N-cadherin in HMCs. (H) Levels of IL-6 and TNF-α in HMCs were measured.
EdU, 5-ethynyl-2'-deoxyuridine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HMC, human mesangial cell; HG, high glucose; IL, interleukin; MEG3, maternally expressed gene 3; miR, microRNA; NC, negative control; OD, optical density; qRT-PCR, quantitative reverse transcription polymerase chain reaction; si, small interfering; TNF, tumor necrosis factor.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
MiR-23c directly targeted LIN28B in human mesangial cells
We predicted the downstream target of miR-23c through the starBase database. As exhibited in Fig. 5A, LIN28B possessed complementary binding sites with miR-23c. We also observed that the luciferase activity of luciferase reporters with LIN28B 3’UTR-WT was reduced in HMCs after co-transfection with miR-23c (Fig. 5B). Moreover, miR-23c and LIN28B enrichment were higher in the anti-Ago2 group (Fig. 5C). Also, LIN28B mRNA levels were also increased in serums from DN patients (Fig. 5D). LIN28B mRNA and miR-23c expression had a negative correlation in serums of DN patients (Fig. 5E). IHC staining also showed the elevated expression of LIN28B in human kidney samples (Supplementary Fig. 6, available online). Analogously, LIN28B protein levels were elevated in HG-induced HMCs (Fig. 5F). These results suggested that LIN28B was a downstream target of miR-23c in HMCs.
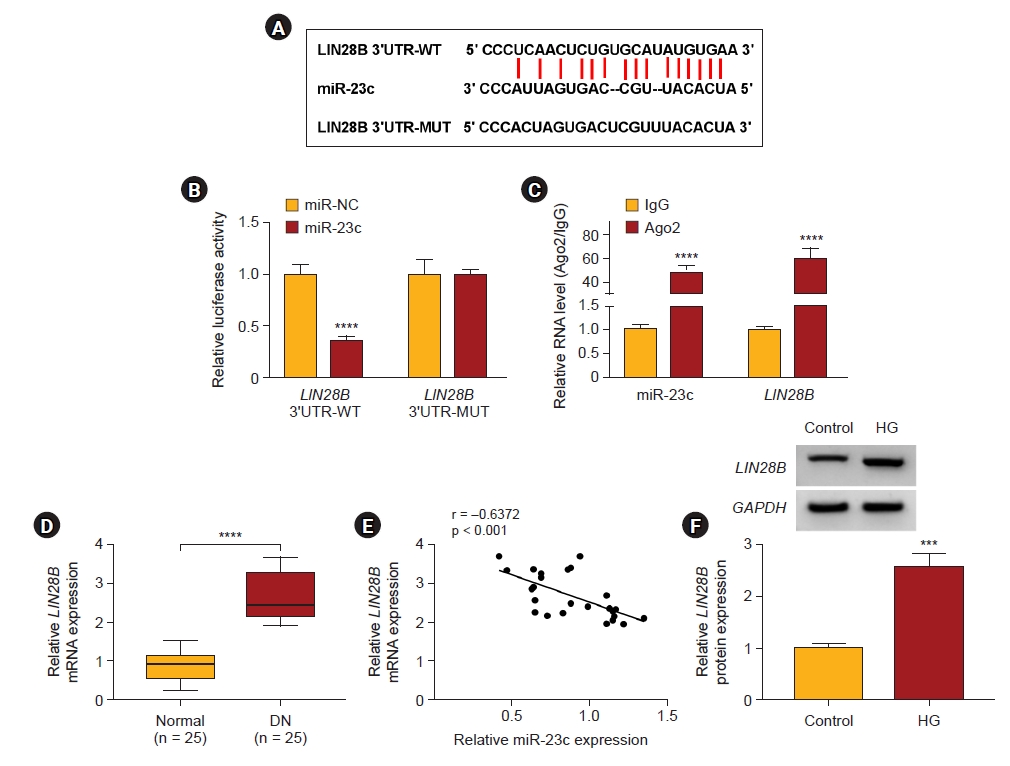
LIN28B served as a target for miR-23c in HMCs.
(A) The diagram exhibited the complementary binding sites between LIN28B and miR-23c. (B) Dual-luciferase reporter assay was executed to check the binding sites between LIN28B and miR-23c. (C) Levels of LIN28B and miR-23c in RNA-induced silencing complexes were examined. (D) Expression of LIN28B mRNA in serums of DN patients. (E) The correlation between LIN28B and miR-23c in serums of DN patients (Pearson correlation analysis). (F) Western blotting analysis of LIN28B protein levels in HG-induced HMCs.
DN, diabetic nephropathy; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HG, high glucose; HMC, human mesangial cell; IgG, immunoglobulin G; LIN28B, lin-28 homolog B; mRNA, messenger RNA; MUT, mutant; miR, microRNA; UTR, untranslated region; WT, wild type.
***p < 0.001 and ****p < 0.0001.
LIN28B overexpression overturned miR-23c mimic-mediated impacts on high glucose-induced human mesangial cell injury
Considering the above results, we further checked whether miR-23c regulated HMCs injury via targeting LIN28B. Western blotting displayed that LIN28B introduction impaired downregulation of LIN28B in HG-stimulated HMCs mediated by miR-23c mimic (Fig. 6A). Also, miR-23c mimic-mediated inhibition of cell proliferation and arrest of cell cycle progression in the G0/G1 phase were reversed by LIN28B elevation in HG-stimulated HMCs (Fig. 6B–E). Furthermore, forcing LIN28B expression overturned the decrease of ECM accumulation, EMT, and inflammation in HG-stimulated HMCs mediated by miR-23c overexpression (Fig. 6F–H). These data indicated that miR-23c modulated HG-induced proliferation, ECM accumulation, and inflammation via targeting LIN28B in HMCs.
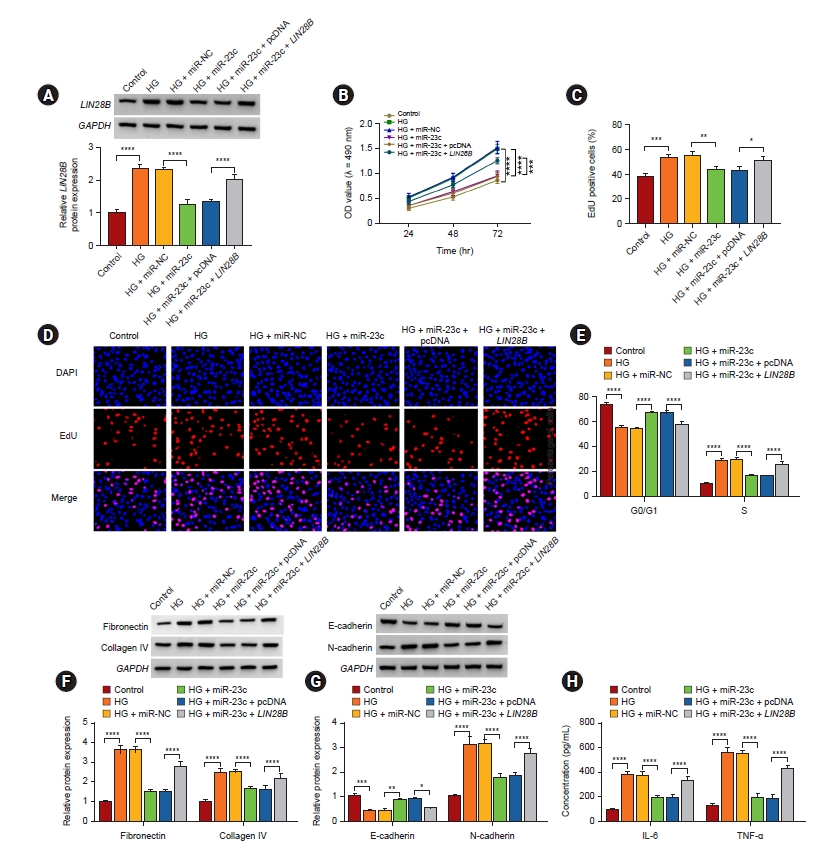
MiR-23c targeted LIN28B to regulate HG-induced HMC injury.
(A–H) After miR-NC, miR-23c, miR-23c + pcDNA, or miR-23c + LIN28B transfection, HMCs were treated with HG. (A) Protein levels of LIN28B protein in HMCs were analyzed. (B–E) The proliferation and cell cycle progression of HMCs were evaluated (D, original magnification of ×200). (F, G) Western blotting presented the levels of fibronectin, collagen IV, E-cadherin, and N-cadherin in HMCs. (H) Levels of IL-6 and TNF-α in HMCs.
EdU, 5-ethynyl-2'-deoxyuridine; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HG, high glucose; HMC, human mesangial cell; IL, interleukin; LIN28B, lin28 homolog B; miRc, microRNA; NC, negative control; OD, optical density; pcDNA, plasmid DNA; TNF, tumor necrosis factor.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
LIN28B was regulated by the MEG3/miR-23c axis in human mesangial cells
Given that lncRNA can function as a ceRNA in the advancement of some diseases, we further analyzed whether MEG3 regulated LIN28B expression via binding to miR-23c. The results exhibited that MEG3 knockdown decreased LIN28B mRNA and protein levels in HG-stimulated HMCs, while this reduction was relieved by miR-23c downregulation (Fig. 7). These data manifested that MEG3 regulated LIN28B expression via binding to miR-23c.
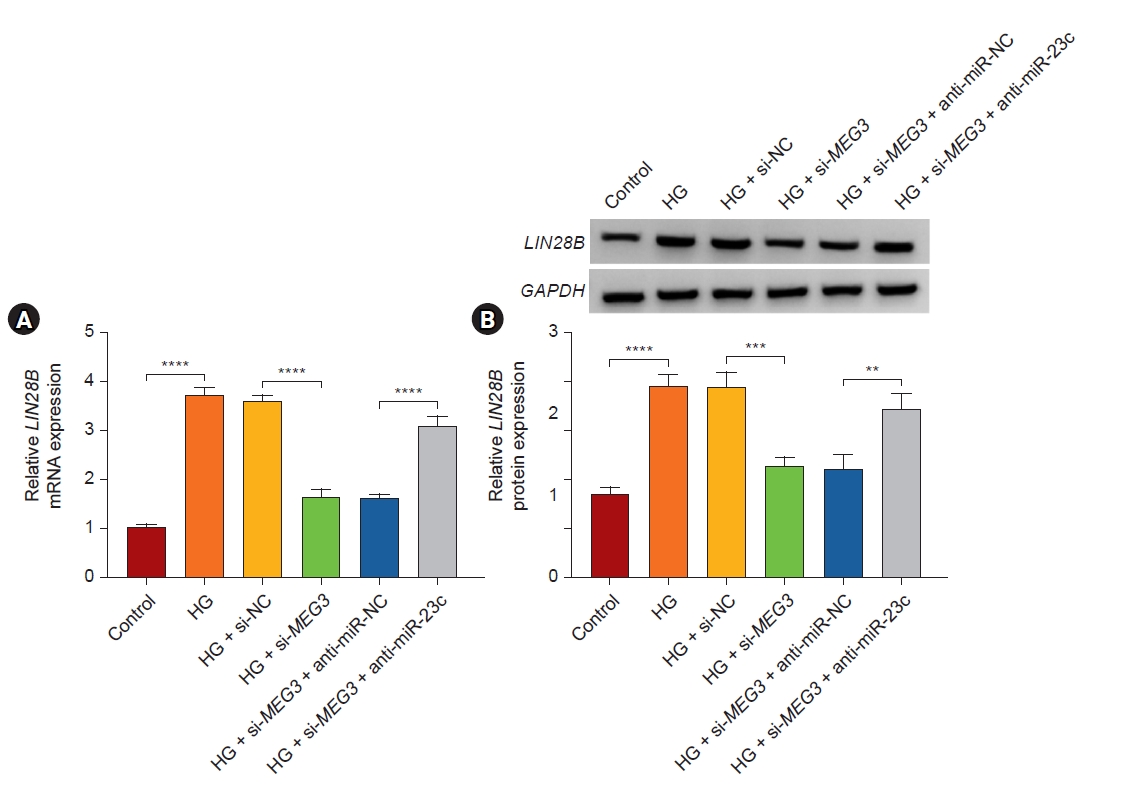
MEG3 modulated LIN28B expression via sponging miR-23c.
(A, B) After HG treatment, LIN28B mRNA and protein levels in HMCs with si-NC, si-MEG3, si-MEG3 + anti-miR-NC, or si-MEG3 + anti-miR-23c were examined.
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HMC, human mesangial cell; HG, high glucose; LIN28B, lin28 homolog B; MEG3, maternally expressed gene 3; miR, microRNA; mRNA, messenger RNA; NC, negative control; si, small interfering.
**p < 0.01, ***p < 0.001, and ****p < 0.0001.
Discussion
Increasing evidence has manifested that lncRNAs can be used as promising therapeutic targets and diagnostic biomarkers for DN [29]. HG is characteristic of DN, which can induce proliferation, EMT, ECM accumulation, and inflammation of HMCs [30,31]. EMT, a phenotypic change process, is accompanied by the loss of E-cadherin and the rise of N-cadherin and is a factor that directly leads to ECM accumulation, especially in DN [32]. Recent studies revealed that MEG3 silencing could induce apoptosis, curb proliferation, and decrease fibrosis of HMCs by sponging miR-145 under HG treatment [21]. Another report uncovered that MEG3 contributed to inflammation and fibrosis of HMCs via regulation of the miR-181a/Egr-1/TLR4 axis [22]. In this study, MEG3 was highly expressed in DN. MEG3 downregulation decreased HMC proliferation, ECM accumulation, and inflammation under HG treatment, which was consistent with the previous studies [21,22], indicating that HG-induced HMC injury relied on MEG3-mediated mechanisms.
More and more evidence showed that lncRNAs regulate gene expression by competitively binding to miRs, thereby participating in the development of various diseases [33]. In DN, lncRNA MALAT1 accelerated pyroptosis of renal tubular epithelial through elevating ELAVL1 expression via sponging miR-23c [27]. Also, lncRNA NEAT1 adsorbed miR-23c in mouse mesangial cells, resulting in facilitating cell proliferation, fibrosis, and EMT [28]. These data indicated that miR-23c played a protective role in the pathogenesis of DN. Our results manifested the sponge action of MEG3 on miR-23c in HMCs. Moreover, miR-23c inhibitor abolished the suppressive impact of MEG3 knockdown on HG-stimulated HMC proliferation, ECM accumulation, and inflammation. Therefore, we inferred that HG-induced MEG3 modulated HMC injury via sponging miR-23c.
LIN28B, an RNA-binding protein, is an important regulator of stem cell maintenance [34] and tumorigenesis [35]. A study revealed that miR-152 could repress HG-induced angiogenesis by targeting LIN28B and inhibiting vascular endothelial growth factor signaling [36]. In DN, transforming growth factor-β1 induced collagen accumulation via the LIN28B/let-7 pathway [37]. Furthermore, miR-379-5p inhibited renal fibrosis through the regulation of the LIN28B/let-7 pathway [38]. Herein, we verified that LIN28B acted as a miR-23c target. LIN28B overexpression overturned the suppressive influence of miR-23c mimic on HG-induced HMC injury. Moreover, MEG3 sponged miR-23c to regulate LIN28B expression. Therefore, we concluded that HG-induced MEG3 regulated proliferation, ECM accumulation, and inflammation via regulating LIN28B expression by sponging miR-23c.
In summary, HG-induced MEG3 adsorbed miR-23c to elevate LIN28B expression, resulting in promoting proliferation, ECM accumulation, and inflammation of HMCs. The study provided a novel mechanism responsible for HG-induced mesangial cell damage.
Supplementary Materials
Supplementary data are available at Kidney Research and Clinical Practice online (https://doi.org/10.23876/j.krcp.23.090).
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Data sharing statement
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
Authors’ contributions
Conceptualization, Methodology: HX, JH
Data curation, Formal analysis: JH, JL, HX
Investigation, Validation: LR, HX
Writing–original draft preparation: LR, HX, JH
Writing–review & editing: LR, HX, JH
All authors read and approved the final manuscript.