



Introduction
Acute kidney injury (AKI) affects 13.3 million people worldwide per year and causes up to 1.7 million deaths annually [1,2]. Regardless of whether renal function recovers fully or not, AKI survivors are at high risk of transitioning to chronic kidney disease (CKD) and, in some cases, progressing to end-stage kidney disease (ESKD). Previously, AKI and CKD were identified as distinct disorders; however, the two syndromes now are considered a continuum of disease progression [3]. Most importantly, some cases do not meet the criteria for AKI or CKD clinically [4]. There is a need to fill this diagnostic gap and optimize post-AKI care, particularly in Taiwan, which has the world’s highest prevalence and incidence of ESKD [5,6].
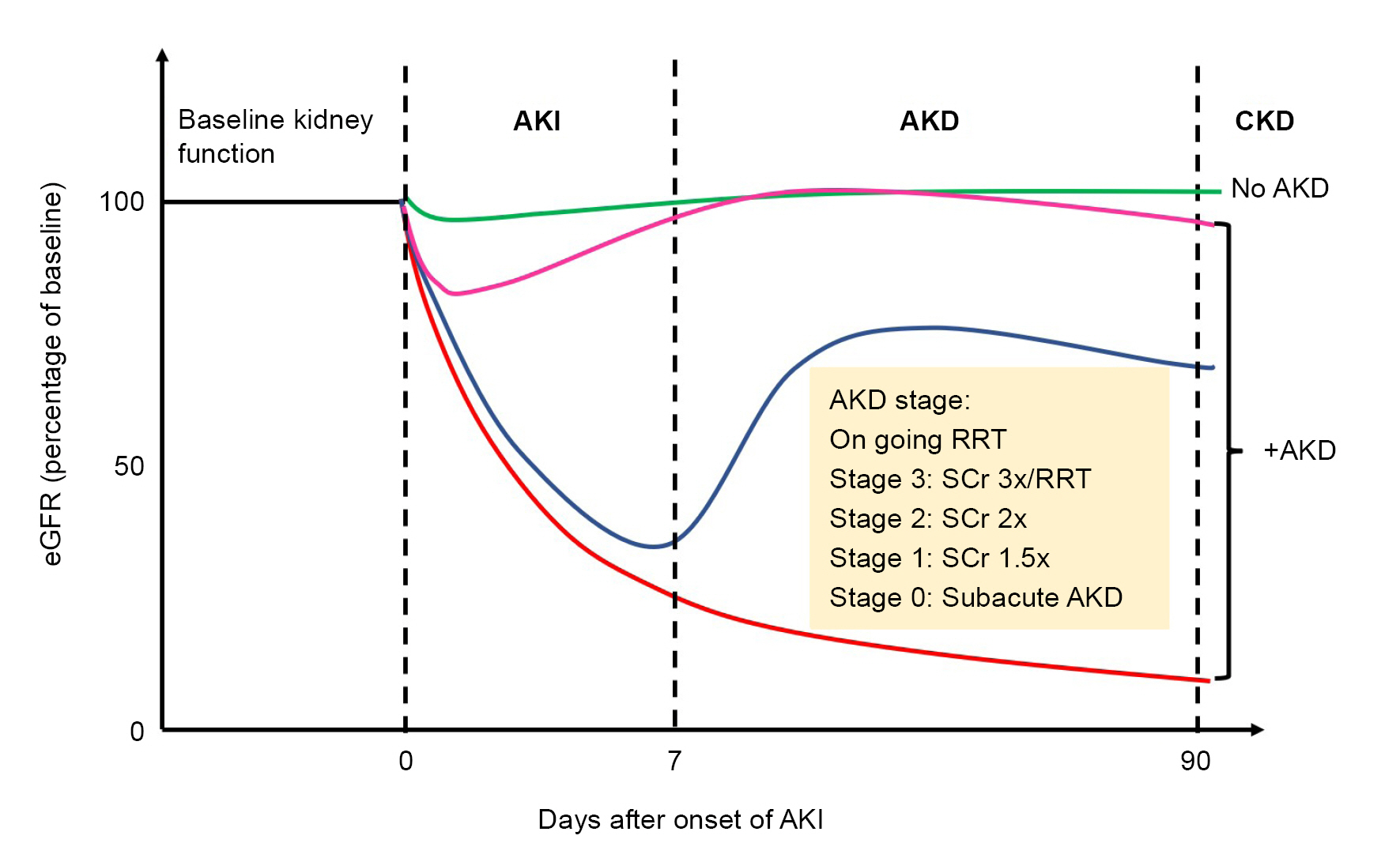
There is a critical phase in the process of the AKI-CKD transition, which is referred to as acute kidney disease (AKD). The concept of AKD was first proposed in the 2012 Kidney Disease Improving Global Outcomes (KDIGO) guideline for AKI. In 2017, the report from the 16th Acute Dialysis Quality Initiative (ADQI) consensus conference defined AKD as subacute damage and/or loss of kidney function, as indicated by changes in serum creatinine or biomarkers occurring 7 to 90 days after AKI and classified as stages 0 to 3 according to the highest serum creatinine level during the AKD period (Fig. 1) [4,7]. This 7- to 90-day period represents the window during which key interventions may be initiated to hinder CKD.
Given that AKD increases the risk of CKD, it is important to enhance awareness of preventive, diagnostic, and therapeutic strategies. We provide a comprehensive review of the literature addressing the epidemiology, pathophysiology, and frameworks for monitoring and treating AKD.
Diagnosis and epidemiology
While AKD is a common sequela of acute illness during hospitalization, it is generally under-recognized; this is problematic because AKD is associated with high morbidity, long-term adverse outcomes, and mortality globally [8]. However, AKD has not been extensively investigated nor have its long-term clinical outcomes. In a large study of 62,977 hospitalized adults with preserved baseline kidney function, See et al. [8] found that AKD occurred in 2.2% and AKI in 7.7% of the patients; 906 (1.4%) had AKD with AKI, and 485 (0.8%) had AKD without AKI. Those who had AKD without AKI had a greater risk of major adverse kidney events (36.21 per 100 person-years; hazard ratio [HR] 2.26, 95% confidence interval [CI] 1.89–2.70), CKD (22.94 per 100 person-years; sub-HR 2.69, 95% CI 2.11–3.43), kidney failure (0.28 per 100 person-years; sub-HR 12.63, 95% CI 1.48–107.64), and death (14.86 per 100 person-years; HR 1.57, 95% CI 1.19–2.07). Patients who were elderly and had more comorbidities were more vulnerable to AKD [8,9].
Additional tools, such as clinical scoring systems, imaging techniques, functional testing, and biomarkers, are needed to diagnose AKD early [3]. Kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) have possible roles as biomarkers for AKD [10–12]. In Cheng et al.’s animal study [13], sustained upregulation of KIM-1 and NGAL was evident 4 weeks after ischemia-reperfusion AKI (IRI-AKI), which was associated with persistent tubular injury and interstitial inflammation. In a clinical study, tissue inhibitor metalloproteinase-2 and insulin-like growth factor-binding protein 7 were found to predict long-term outcomes of AKI patients [14]. In a recent single-center study of 430 patients with community-acquired and hospital-acquired AKI, concurrent clinical evaluation with “risk scoring” and urine sediment analysis was reported to be a promising method to predict the AKI-CKD transition [15]. Elevated urinary angiotensinogen is also considered a strong predictor of AKI progression [16]. To improve diagnosis and risk stratification after a hospitalized episode of AKI, an ongoing multicenter study (ASSESS-AKI study) is investigating differences in the occurrence of renal and cardiovascular outcomes and death within a cohort of patients with or without AKI. This study will also evaluate the efficacy of urine biomarkers (NGAL, KIM-1, cystatin C, interleukin [IL]-18, L-type fatty-acid-binding protein, and N-acetyl-β-d-glucosaminidase) and blood biomarkers (serum NGAL, serum cystatin C, and plasma IL-6) [17]. Several studies have been performed using the database of the ASSESS-AKI study. First, Wen et al. [18] found that adjusting urine IL-18, urine KIM-1, and monocyte chemoattractant protein-1 (MCP-1) concentrations for urine creatinine or urine osmolarity in patients with AKI could strengthen predictions of subsequent CKD. In addition, when the biomarkers assessed in the ASSESS-AKI study were applied in a mouse model of IRI-AKI, greater MCP-1 and YKL-40 concentrations were associated with greater reduction of eGFR and increased incidence of the composite renal outcome, whereas greater uromodulin concentration was associated with lower incidence of the composite kidney outcome [19]. Second, Coca et al. [20] reported that plasma soluble tumor necrosis factor receptor 1 (sTNFR1) and sTNFR2 measured 3 months after hospital discharge were independently associated with kidney disease progression, heart failure, and mortality regardless of AKI status. Another study conducted by Mansour et al. [21] found that greater angiopoietin-1 to angiopoietin-2 ratio was strongly associated with less frequent CKD progression, heart failure, and mortality. We look forward to using these novel biomarkers to predict and diagnose AKD in the future and propose that a multimarker score be used to improve risk stratification of patients and to increase prognostic accuracy.
Moreover, a meta-analysis of nine studies suggested that an elevated Doppler-based renal resistive index was an accurate predictor of persistent AK I in critically ill patients [22]. Chawla and Ronco [23] proposed that noninvasive kidney stress testing, an approach using appropriate stimulants to assess reserve capacity in the kidney, could be used to monitor renal function and improve prognostic predictions. The abovementioned biomarkers and detection methods may allow more timely diagnosis and earlier intervention than conventional renal function tests, such as serum creatinine concentration [24].
Risk factors
Several complications may occur in AKD patients, including cardiovascular events, diminished quality of life, development of disability, and increased mortality [25]. Many patient characteristics are risk factors for AKD, such as age, race, genetic factors, hypertension, diabetes mellitus, and metabolic syndrome [3]. In addition, the severity, duration, and frequency of AKI are independent risk factors for CKD. Kellum et al. [26] found that the severity of AKI, as classified by the KDIGO criteria, was a powerful predictor of progression to CKD. Thakar et al. [27] assessed 3,679 diabetic patients and found that AKI episodes were associated with a cumulative risk of progressive CKD.
Advanced age, as well as history of surgery, coronary angiography, coronavirus disease 2019 (COVID-19) infection, fluid overload, and some pharmacological agents, such as chemotherapeutic agents and the frequently used nonsteroidal anti-inflammatory drugs (NSAIDs), are modifiable factors that contribute to AKD [28–32]. Chou et al. [33] conducted a single-center cohort study of 6,101 patients with stage 3B-5 CKD from 2005 to 2018 and concluded that older patients were more vulnerable to AKI and progression to ESKD compared with younger patients. Ishani et al. [34] assessed 29,388 patients undergoing cardiac surgery and found that increased risk of subsequent CKD could be predicted by an increase in serum creatinine after cardiac surgery, even in those with mild severity. Among 11,249 Canadian residents aged 18 years or older undergoing coronary angiography, James et al. [30] discovered a relationship of AKI after coronary angiography with an elevated risk of sustained renal function loss. Notably, in a recent cohort study of 1,017 COVID-19 patients who were ever admitted to a hospital but survived until the day of discharge, Hadadi et al. [32] reported a possible long-term influence of COVID-19-associated AKI on the persistence of renal dysfunction.
In a large observational study performed over five centers across the United States that included patients from different demographic groups, Bouchard et al. [28] analyzed 610 critically ill patients who had ever consulted a specialist for AKI occurring in the intensive care unit. They found that fluid overload at the time of diagnosis of AKI, defined as a greater than 10% increase in body weight relative to baseline, was not related to kidney function recovery. However, fewer patients in whom peak serum creatinine concentration occurred concurrently with fluid overload recovered kidney function compared with those who did not have these factors (35% vs. 52%, p = 0.007) [28]. On the other hand, AKI and CKD can both give rise to and result from malignancy. Some traditional chemotherapeutic agents are nephrotoxic and can aggravate kidney dysfunction, and impairment of renal function also appears following the use of some newly developed chemotherapeutic agents. Proper diagnosis and management are required to reduce chemotherapy-related renal toxicity [31].
Mechanisms
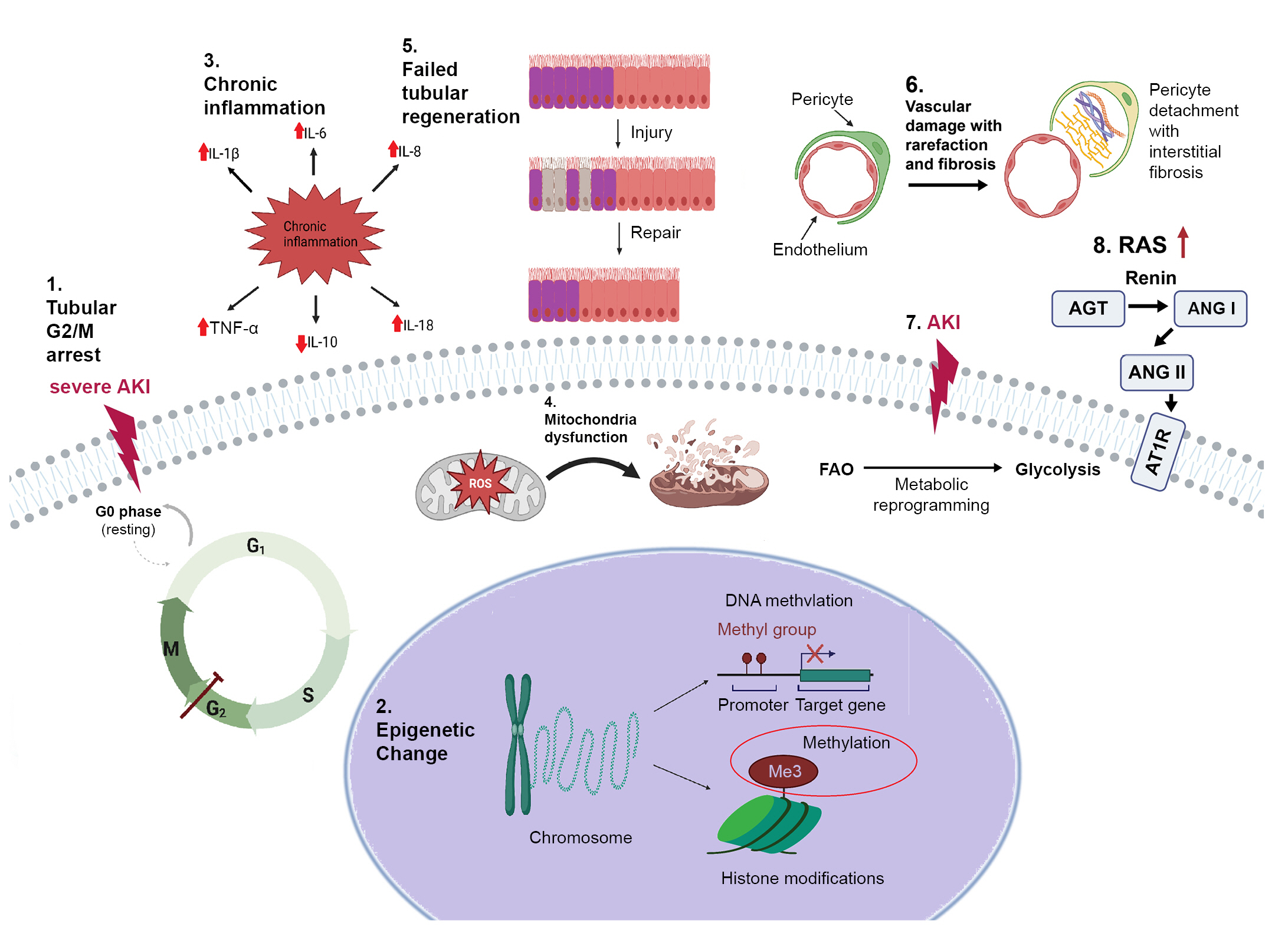
Several plausible mechanisms leading to AKD have been proposed, including renal tubular epithelium cell-cycle arrest, epigenetic changes, failure to recover from inflammation after AKI, mitochondrial dysfunction, failed regeneration of proximal tubules, endothelial dysfunction, metabolic reprogramming, and activation of the renin-angiotensin system (RAS) (Fig. 2). Novel monitoring strategies and therapeutic agents could be developed from investigations of these mechanisms.
Renal tubular epithelium cell-cycle arrest
A recent study demonstrated G2/M cell-cycle arrest of renal tubular epithelial cells (TECs) as a key factor contributing to maladaptive repair and progression to CKD after AKI [35]. Notably, in these G2/M-arrested tubular epithelium cells, activated c-jun NH2-terminal kinase signaling can boost the production of profibrogenic growth factors such as transforming growth factor-β1 (TGF-β1) and connective tissue growth factor. While TGF-β1 can lead to cell apoptosis, it also reinforces tubular cell arrest in the G2/M phase. Elevated production of profibrogenic cytokines activated the generation and transformation of pericytes to myofibroblasts and enhanced advanced fibrosis in a murine obstructive AKI model [36].
Epigenetic change
Epigenetic change, an inheritable gene expression change not caused by alterations in the primary nucleotide sequence, was first proposed by Waddington in 1942 [37]. Epigenetic regulation of gene expression is the result of DNA methylation, histone acetylation/deacetylation, and microRNA (miRNA) expression. Bechtel et al. [38] demonstrated that RASAL1 is a gene encoding the RAS oncoprotein inhibitor. Hypermethylation of RASAL1 induces fibroblast activation, proliferation of fibroblasts, and renal fibrogenesis following folic acid-induced AKI, leading to the CKD transition. Kidney fibrosis is ameliorated by the demethylating agent 5-azacytidine which acts upon the methyltransferase Dnmt1 [38]. Recently, Chou et al. [39] reported that demethylation by 5-azacytidine could restore the microvascular stabilization of activated pericytes and reverse the profibrotic features of inactivated pericytes, preventing the transition to CKD and attenuating fibrogenesis. In conclusion, blockage of hypermethylation in pericytes after AKI could block the transition to CKD.
Alternation of histone expression is also involved in the mechanism of AKD. Two major gene-activating histone modifications (histone 3 lysine 4 trimethylation and histone variant H2A.Z) have been demonstrated in a murine model of IRI-AKI. These changes increase the production of renal cortical TGF-1/MCP-1 cytokines and collagen deposition [40].
Multiple miRNAs are involved in epigenetic changes after AKI, especially miRNA-24, miRNA-494, miRNA-21, miRNA-127, and miRNA-687 [41]. Specifically, miRNA-24 stimulates endothelial and TEC apoptosis, leading to renal ischemic injury. After AKI, upregulated miRNA-494 leads to inflammation or adhesion-molecule-induced kidney injury by inhibiting the expression of activating transcription factor 3. Upregulation of miRNA-21 protects against AKI through interacting with miRNA-21 and hypoxia-inducible factor (HIF); miRNA-127 likewise protects against IRI-AKI through HIF-1. The PTEN gene is inhibited by miRNA-687, facilitating renal tubular epithelium cell-cycle activation for proliferation.
Chronic inflammation
Inflammation can develop during an episode of AKI and persist thereafter. Initially, resident dendritic cells and macrophages increase in response to cell injury to release chemotactic signals that recruit leukocytes to the kidney [42]. A recent study in an aged mouse model showed that impaired M2 macrophage polarization with cell-cycle arrest might accelerate progression from AKI to CKD. During the recovery phase in aged mice, the expression of cell-cycle arrest markers was increased in renal TECs in the G1 phase. Mononuclear cells and M1 macrophages have impaired M2 polarization in vitro. This finding indicates that persistent M1 inflammation may result from prolonged G1 arrest in aged mice. Finally, fibrosis is developed in aged mice due to M1-dominant inflammation [42]. These findings may underlie the phenomenon of higher risk of CKD after AKI among elderly patients.
With regard to the evolution of renal injury, Cheng et al. [13] showed that B and T cells infiltrate and persist despite functional recovery even 28 days after IRI-AKI in mice. Moreover, tertiary lymphoid tissues (TLTs), which are the site of origin of local immune responses and play an essential role in chronic inflammation, have been found in aged mice after AKI and in elderly patients with several diseases [43]. However, the associations of TLTs with renal function progression are unclear. Therefore, Sato et al. [44] analyzed kidney samples from elderly patients and patients with pyelonephritis and found that the stage of TLT could reflect local injury and inflammation. Furthermore, in a murine model of IRI-AKI, deletion of CD4+ cells and late administration of dexamethasone were accompanied by a reduction in TLT stage and further improvement of renal function, fibrosis, and inflammation.
Other emerging studies have shown that salt-inducible kinase 1 (SIK1) plays an essential role in the AKI-CKD transition. Hu et al. [45] reported that SIK1 was down-regulated in AKI and could induce the AKI-CKD transition through activation of the WNT/β-catenin signaling pathway. Overexpression of SIK1 alleviated the AKI-CKD transition in vivo and renal tubular cellular injury in vitro.
Mitochondrial dysfunction
Recent findings have revealed that fragmented mitochondria induce persistent renal injury by releasing harmful molecules that can activate nucleotide-binding oligomerization domain-like receptors and increase the release of proinflammatory cytokines. Mitochondrial damage persists long enough after ischemic renal injury to activate sustained chronic inflammation, which can cause continued endothelial and podocyte damage and microvascular rarefaction, ultimately leading to glomerular and interstitial fibrosis. In addition, under oxidative stress, a damaged mitochondrial membrane leads to the release of reactive oxygen species and proapoptotic factors, such as cytochrome complex and apoptosis-inducing factor; apoptosis is therefore enhanced, and renal recovery from AKI is impaired [46].
Failed regeneration of tubular cells
Some surviving dedifferentiated tubules fail to undergo regeneration after AKI. Endo et al. [47] demonstrated that acute tubular injury caused significant shortening of proximal tubules and was associated with limited repair and subsequent interstitial fibrosis. Failure of tubular regeneration leads to reduced renal mass, which results in increased intraglomerular pressure and glomerular hyperfiltration [13,25]. Loss of parenchyma following renal mass reduction elicits hemodynamically mediated processes that give rise to glomerulosclerosis and tubulointerstitial fibrosis [48].
Endothelial dysfunction
Microvascular rarefaction is thought to play a crucial role in nephron damage and has been associated with progression to chronic nephropathy. Basile et al. [49] found loss of capillaries even tubular morphology was essentially normal at 4 and 8 weeks after IRI-AKI in a murine model, indicating deficient regenerative potential of the renal vascular system. Microvascular rarefaction-induced renal hypoxia leads to mitochondrial dysfunction, proinflammatory induction, and profibrogenic processes [50]. Epithelial cells produce an endogenous cytokine named vascular endothelial growth factor, which affects endothelial cells directly and is reduced after AKI. Therefore, microvascular dysfunction and morphologic changes are induced in the nephron upon AKI [51,52]. In addition, renal pericytes are widely recognized to play a key role in microvascular stability. Following AKI, pericytes detach from capillaries and then differentiate into myofibroblasts. The loss of pericytes leads to transient injury of the renal tubules and long-term rarefaction of peritubular capillaries, which increases the risk of progression to CKD after AKI [53]. Loss of capillaries induces hypoxia, which stimulates production of fibrogenic factors including TGF-β1 and extracellular matrix [54,55]. A vicious cycle ensues, such that the larger is the number of interstitial scars, the longer is the distance for diffusion to the renal parenchyma, which aggravates hypoxia and renal fibrosis [56,57]. Yamaguchi et al. [58] also demonstrated that hypoxia may induce inflammation by causing overexpression of CCAAT/enhancer-binding protein δ through nuclear factor-κB-dependent pathways.
Metabolic reprogramming
During AKI, reprogramming of energy metabolism occurs in TECs due to hypoxia, mitochondrial dysfunction, and disordered nutrient-sensing pathways. Fatty acid transporters including CD36 and fatty-acid-binding proteins as well as fatty acid β-oxidation (FAO)-related metabolic enzymes such as carnitine palmitoyltransferase I and medium chain-specific acyl-CoA dehydrogenase are reduced in TECs [59]. Therefore, the metabolic process switches from FAO to glycolysis. Enhanced glycolysis provides the energy supply; however, in the long-term, glycolysis leads to inflammation, lipid accumulation, and fibrosis, which promote the AKI-CKD transition [60].
Renin-angiotensin system activation
An increasing number of studies have demonstrated activation of the RAS after AKI. In cardiac surgery-associated AKI (CSA-AKI), low blood flow, low pressure, hypothermia, and neurohormonal factors related to surgery are risks for AKI because of increased renal vasoconstriction due to RAS activation [61]. In addition, RAS activation in AKI patients is proportional to the severity of AKI and the level of urinary angiotensinogen [62].
Hsu et al. [63] conducted a retrospective cohort study that demonstrated an independent association between AKI and subsequent development of elevated blood pressure. In vivo, Cheng et al. [13] demonstrated a link between blood pressure elevation/intrarenal RAS activation and CKD. Losartan treatment initiated 1 month after functional recovery from AKI significantly reduced the rate of subsequent CKD and mortality. Clinically, Chou et al. [64] analyzed a cohort study retrospectively and confirmed a greater decrease in the rate of subsequent CKD (users vs. nonusers, 26.6% vs. 42.2%) and a longer median CKD-free survival time (users vs. nonusers, 1,079 days vs. 520 days) in patients who initiated and maintained compliance with RAS blockade after full renal recovery from CSA-AKI.
However, RAS blockade is not usually initiated during the acute stage of AKI and should be used cautiously. It also has been reported that AKI admission rates are associated with increased prescriptions of RAS inhibitors [65].
Treatment and prevention
Prevention of repeated acute kidney injury
The best way to prevent AKD is to decrease the occurrence of AKI [66]. A number of studies have focused on this topic [67,68]. However, once an episode of AKI occurs, it becomes crucial to prevent the transition to CKD and improve AKI recovery. This can be achieved through the use of promising new pharmacological agents or the development of innovative therapies, such as stem cell therapy. [69,70] to attenuate the risk of CKD. A meta-analysis of four randomized controlled trials and nine non-randomized studies that included 25,776 patients and 30,276 AKI episodes found that AKI care bundles reduced the rate of moderate to severe AKI [71]; however, it is unclear whether this improvement translates into better long-term outcomes.
Early and regular follow-ups by a nephrologist
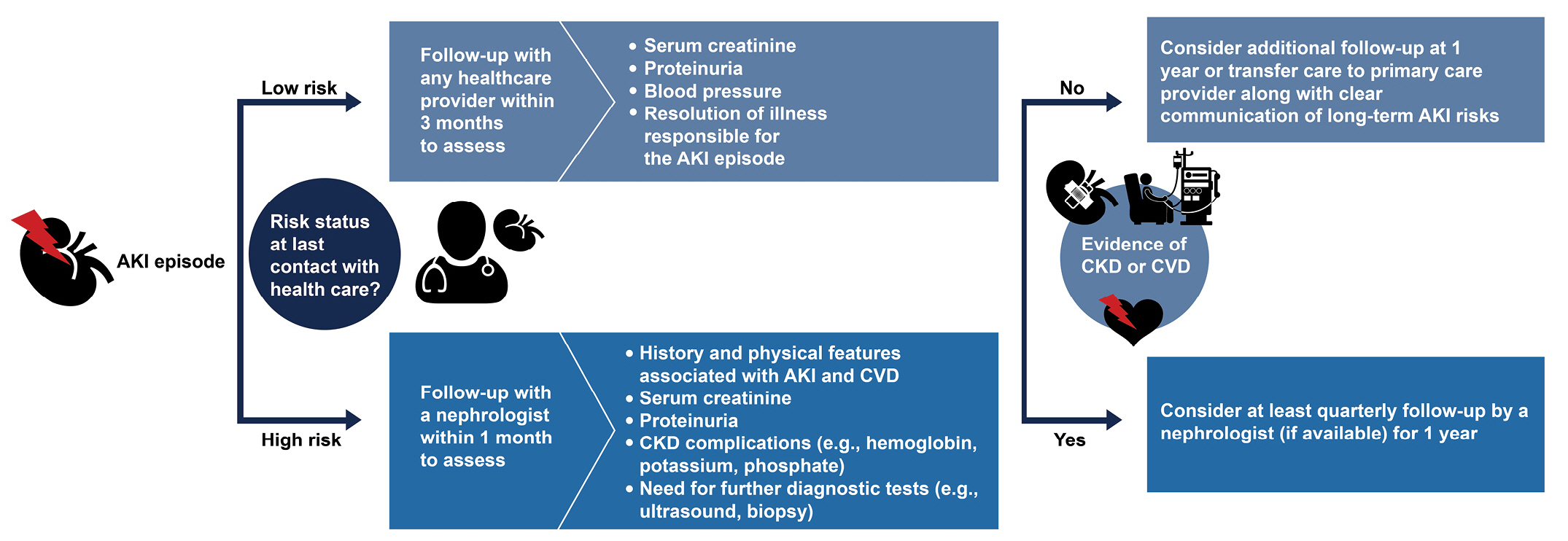
Studies reporting observations of AKI survivors are rare, and the lack of prospective observational studies that have followed discharged AKI survivors to obtain long-term outcomes may lead to over- or underestimation of CKD evolution. This lack of data may reflect hospitalization-related fatigue, unwillingness to add more doctors to outpatient care, and long travel distances to medical centers [71]. This data gap suggests a missed opportunity to block the AKI-CKD transition and improve outcomes [72]. Siew et al. [73] reported that 3,929 survivors of AKI were hospitalized between January 2003 and December 2008, as recorded in the United States Department of Veterans Affairs database, which demonstrated that AKI severity did not affect referral rates, but only a few high-risk survivors were transferred to nephrologists. However, it has been shown that early nephrology follow-up has many benefits, such as timely interventions, improved outcomes, better access to medical resources, and more consistent follow-up. A cohort study showed that early nephrologist follow-up, meaning a visit with a nephrologist within 90 days of discharge, reduced all-cause mortality [74]. However, there is disparity between what nephrologists consider ideal care after discharge and the transitional care currently provided in practice. Some Canadian nephrologists demonstrated in 2015 that 16% of AKI patients without preexisting CKD visited a nephrologist at 6 months after AKI, whereas this frequency increased to 24% among AKI survivors who had preexisting CKD and had been visited by a nephrologist before or during their AKI hospitalization period (78% and 41%, respectively) [75].
Resume and adjust essential medications
Certain medications are withdrawn or adjusted during an episode of AKI, such as NSAIDs, diuretics, RAS inhibitors, statins, and oral hypoglycemic agents. In addition, the combination of spironolactone, NSAIDs, and RAS inhibitors should be avoided. Although ‘sick day medication guidance’ has been applied in clinical practice for patients with diabetes mellitus and CKD, it has not yet been affirmed that withdrawal of medications such as angiotensin-converting enzyme inhibitors (ACEi) and/or angiotensin receptor blockers (ARBs) could prevent adverse events, including AKI. The timing of drug reinitiation needs to consider the degree of post-AKI recovery, the interval to AKI insult, the drug’s metabolic and excretion pathways, indications for the drug, and whether there is any suitable substitute [3]. After the introduction of ACEi/ARB, spironolactone, trimethoprim, and loop diuretics (especially for patients with preexisting CKD), renal function should be monitored closely for a brief period. Finally, drug choice, dosing, and surveillance during the AKI-CKD transition must be emphasized, and nephrotoxic medicines should be avoided [7].
Optimize blood pressure control and nutrition management
A large retrospective cohort study of adult patients who were hospitalized between 2008 and 2011 (after exclusion of those who had at least two blood pressure readings greater than 140/90 mmHg or those with evidence of CKD) found that AKI was independently associated with subsequent development of elevated blood pressure [63]. Accordingly, it is worth investigating how to optimize blood pressure control after AKI.
Baek et al. [76] conducted a retrospective cohort study including 1,612 noncritically ill hospitalized patients with AKI and found a U-shaped curve between the average systolic blood pressure (SBP) within 48 hours after AKI and AKI severity or 90-day mortality. The lowest event rate occurred when SBP was controlled to within approximately 110 and 129 mmHg. In contrast, another prospective study found the inverse relationship. McCoy et al. [77] found that SBP 3 months after discharge was not associated with the risk of AKI, loss of kidney function, mortality, or heart failure events among hospitalized AKI patients. In addition, a Japanese study that enrolled 746 patients admitted to intensive care units after cardiac surgery demonstrated no association between hypotension and subsequent progression of AKI [78]. In view of the aforementioned studies, the optimal strategy for post-AKI blood pressure control to prevent ensuing CKD requires additional randomized controlled trials for validation.
After an episode of AKI and during recovery, patients are at risk of protein-energy malnutrition, and this is not ideal for a good prognosis. Renal function loss affects the metabolism of all macronutrients, and hypertriglyceridemia and hyperglycemia are common in this situation. It is necessary to individualize AKI survivor nutrition support based on etiology, severity, comorbidities, and dialysis demands [79]. Fiaccadori et al. [80] suggested in 2010 that AKI patients requiring dialysis should take in at least 1.5 g/kg/day of protein with an extra 0.2 g/kg/day due to dialysis-related losses. No more than 30 kcal/kg/day of nonprotein calories or 1.3 × basal energy expenditure calculated by the Harris-Benedict equation, with 30% to 35% from lipids, should be considered in the energy intake. The use of the gut (oral or enteral support) is favored [80]. A recent original article using evidence-based medicine methods to review the medical literature for Taiwanese AKI patients from 2014 to 2018 also supported that suggestion [81].
For fluid requirements, it is not wise to apply standard fluid equations. Close medical assessment based on the patient’s fluid balance is crucial [77]. Electrolytes should be carefully monitored and adjusted, especially food containing a high level of potassium. Requirements for micronutrients are not well documented. Whether micronutrient supplementation improves outcomes remains unknown [82]. However, Fiaccadori et al. [83] pointed out that the recent guideline was aimed at providing evidence-based nutritional recommendations for different clinical settings of hospitalized patients.
Potential pharmaceutical agents to prevent the acute kidney injury-chronic kidney disease continuum
To promote AKI recovery, Silver et al. [84] suggested clinicians in the care of AKI survivors consider lifestyle modifications, medication adjustment, blood pressure control, medical record documentation, and patient education in their management plan. Moreover, individualized care should be provided. In addition, glycemic control for diabetic patients, RAS blockade, and statins could reduce the risks of adverse long-term cardiovascular and renal outcomes after AKI [9]. Interestingly, some bioactive compounds, such as growth factors and cytokines, have shown effectiveness in facilitating kidney function recovery [85].
Because RAS activation is one of the mechanisms of AKD, RAS inhibitors could become robust and safe pharmacological agents for blocking the AKI-CKD transition [13,69,86]. One animal study and another clinical study showed that RAS inhibitors could reduce post-AKI mortality and progression to CKD [13,64]. Moreover, an animal study also demonstrated that treatment with spironolactone, an antagonist of the downstream RAS effector aldosterone, either before or after ischemia, prevented subsequent CKD [87].
Chou et al. [39] reported a breakthrough experimental result that, in 5-azacytidine–treated mice, the number of myofibroblasts and severity of fibrosis decreased along with decreased blood urea nitrogen and creatinine concentrations on day 180 after AKI compared with the control group. These findings suggest that demethylation by 5-azacytidine attenuates the AKI-CKD continuum; however, more clinical trials are needed to investigate the effects of demethylating agents in clinical applications.
Current guidelines suggest statin therapy for CKD patients aged over 50 years or with known coronary disease, diabetes mellitus, or prior ischemic stroke or at high cardiovascular risk. A nationwide cohort study found that statins used in patients who were first hospitalized because of AKI requiring dialysis (AKI-D) led to a lower risk (21%) of 1-year all-cause mortality [88]. The survival benefit was dose-dependent, compliance-dependent, sustainable, and similar across patient subgroups. Interestingly, Brar et al. [89] also reported that statin use reduced mortality and rehospitalization rates in AKI survivors who subsequently developed CKD. Based on the current findings and given the not yet established guidelines for statin use in AKI individuals, it is accepted that patients with AKI-D should not discontinue using statins. In the future, more studies must confirm these results and verify the role of statin therapy in the optimal care of AKI patients.
Dipeptidyl peptidase-4 inhibitor (DPP-4 inhibitor), a widely used antihyperglycemic medication for type 2 diabetic control, has also shown some efficacy in reducing ESKD and mortality in type 2 diabetic patients recovering from AKI with dialysis [90]. The potential renal effect of DPP-4 inhibitor may offer resources to enhance success rates in blocking the AKI to CKD transition.
Summary and perspective
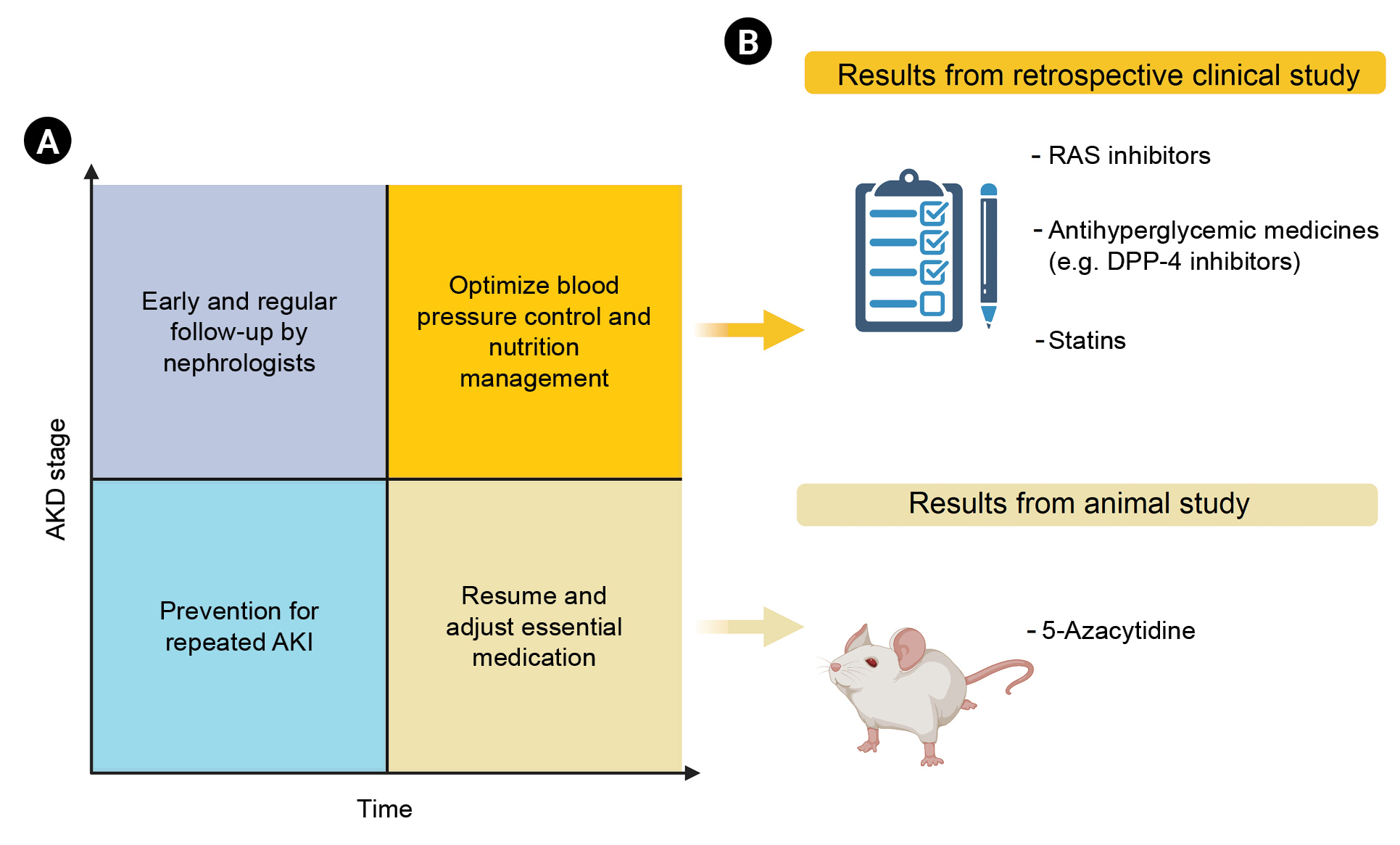
To date, no reliable trials have assessed which are the most powerful pharmacotherapies for treating AKD. Additional investigation of the etiology and underlying mechanisms of AKI progression to AKD are needed to guide the development of effective treatments. The current gold standard for AKD prevention includes identifying at-risk patients, reviewing medical records, and minimizing nephrotoxic exposure. In addition, it is worth emphasizing the value of the clinical utilization of newly identified biomarkers and close follow-up by nephrologists. Therefore, we are proposing a care bundle for AKD based on important lessons from available studies and registries, as shown in Fig. 3, and list monitoring strategies and potential therapeutical agents of AKD in Fig. 4. Considering the significant prognostic impact of AKD, it is crucial to initiate randomized, controlled clinical trials of strategies for AKD prevention and therapy.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









