



Introduction
Fabry disease (FD) is a recessive X-linked lysosomal storage disorder caused by the deficient activity of α-galactosidase A (α-Gal A) enzyme as a result of mutations in the GLA gene. The enzyme deficiency results in the progressive accumulation of glycolipids, specifically globotriaosylceramide (GL-3) and its derivatives such as globotriaosylsphingosine (lyso-GL3), in the vascular endothelium and other tissues. This leads to damage in multiple organs, including kidneys, heart, and cerebrovascular system, and ultimately, premature death.
It is difficult to recognize patients with FD due to its rarity and nonspecific symptoms. Certain treatment strategies, such as enzyme replacement therapy (ERT), can attenuate the disease progression of FD. Hence, it is crucial to screen high-risk populations, such as patients with chronic kidney disease (CKD). Screening of families of patients with FD provides an opportunity for early diagnosis and treatment of FD. The European Renal Best Practice group has recommended FD screening for CKD patients with an unknown etiology [1]. Several screening studies have been performed outside of Korea, with the majority of patients being either male or on dialysis. Additionally, most of the studies have screened FD only via an α-Gal A enzyme test, even though plasma lyso-GL3 has been considered a more useful biomarker. There also have been a few screening studies in Korea, but those were performed in cardiology patients or used serum GL-3 in patients treated on dialysis [2â4]. Therefore, this is the first study in Korea that evaluated the prevalence of FD in adult patients with CKD including those on renal replacement therapy (RRT) by means of two screening tests, α-Gal A enzyme activity and level of lyso-GL3. In addition, the study included both male and female patients, as well as patients with CKD.
Methods
Study design
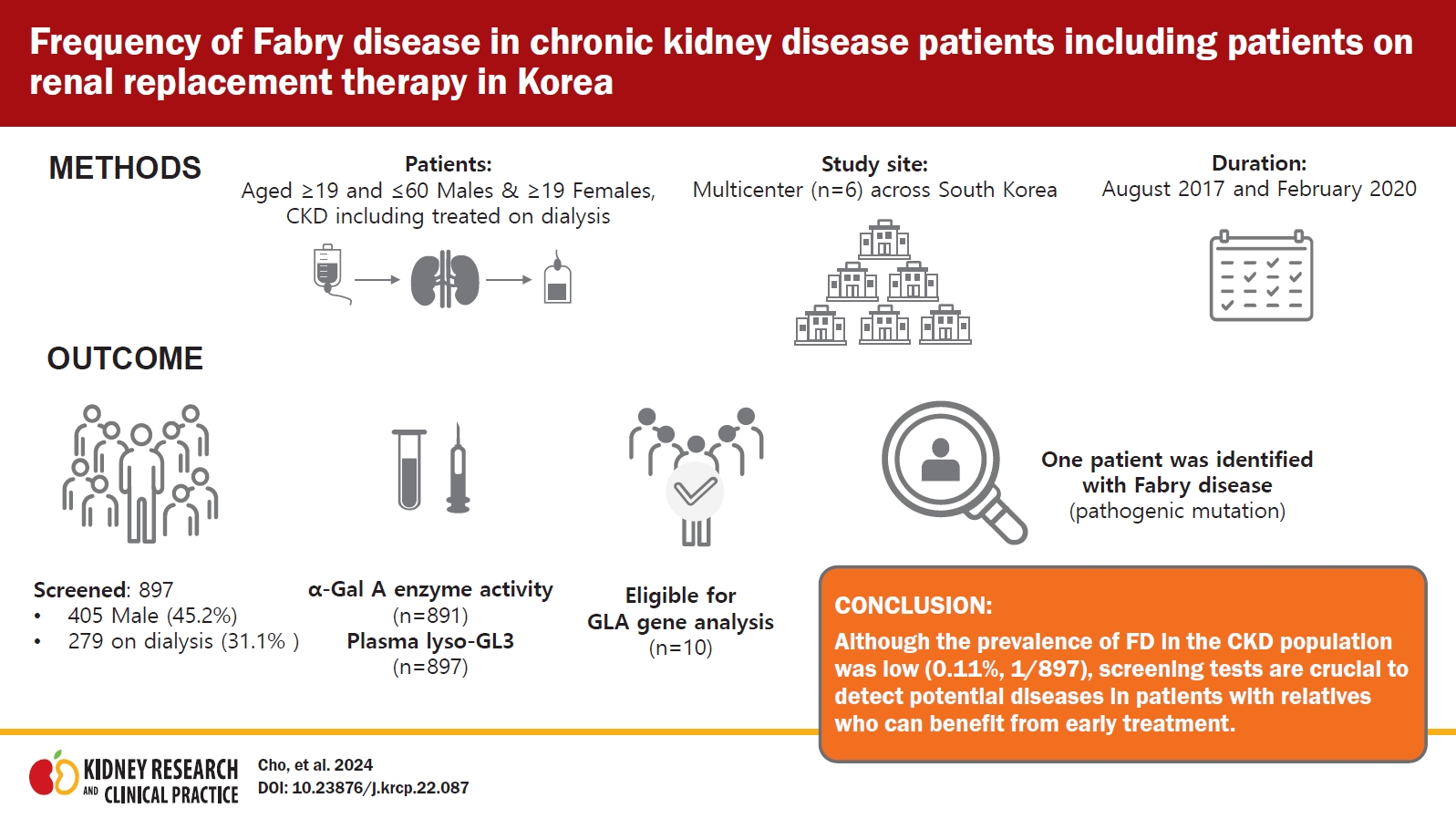
This was a national, multicenter, observational study conducted from August 24, 2017 to February 28, 2020 using a cross-sectional and retrospective study design. Six sites were selected considering the geography, urban/rural practice, and regional population size of South Korea. All centers were university hospitals; four located in Seoul, and one respectively in Busan and Gwangju. All investigators were nephrologists who were instructed to enroll consecutive patients in the study to prevent potential bias during selection of the patients.
Both outpatients and inpatients were recruited if they met the following inclusion criteria. Male patients aged between â¥19 and â€60 years and female patients aged â¥19 years, who were on dialysis, or whose urine protein creatinine ratio (PCR) was â¥150 mg/g or urine albumin creatinine ratio (ACR) was â¥30 mg/g in two consecutive tests conducted during different visits, were included. With regard to CKD, only the presence of albuminuria or proteinuria was considered, taking into account that albuminuria/proteinuria is the early sign of Fabry nephropathy [5,6]. Kidney transplant patients were also included if their pretransplant condition met the inclusion criteria. For patients retrospectively enrolled in this study, their diagnostic tests must have been performed after January 1, 2016. Patients with confirmed etiology on kidney biopsy or who were considered to have typical diabetic nephropathy, as determined by the responsible investigator, were excluded.
After informed consent was obtained, blood sample was collected and transported directly to Seoul Clinical Laboratories. Screening for FD was performed by measuring both α-Gal A enzyme activity and plasma lyso-GL3 concentration before conducting the GLA gene test (Fig. 1). Patients with α-Gal A enzyme activity of â€35 nmol/hr/mg protein in whole blood or â€2.35 µmol/hr/L on a dried blood spot (DBS) test, or with lyso-GL3 of >1.74 ng/mL in plasma, were eligible to undergo a GLA gene test to confirm the diagnosis of FD. Informed consent for gene test was obtained when a patient had abnormal results in screening tests. A single blood sample collection for screening was used for all required tests to minimize patient stress and the risk of missing data. All laboratory tests were performed at Seoul Clinical Laboratories.
Demographic data (age, sex, and ethnicity) and the type of RRT were recorded. Clinical and laboratory data, if available, were collected in the patient records. FD-related symptoms were also collected as follows: 1) cardiac dysfunction including angina, arrhythmia, congestive heart failure, left ventricular hypertrophy, and myocardial infarction; 2) neurologic symptoms including pain, depression, and anxiety; 3) ocular symptoms including corneal and lenticular opacities and vasculopathy; 4) pulmonary symptoms including airway obstruction; 5) gastrointestinal symptoms including dyspepsia; 6) ears, nose, and throat (ENT) symptoms including hearing loss; 7) dermatologic symptoms.
This trial was registered with the Korean Research-based Pharma Industry Association (https://www.krpia.or.kr/eng); the unique identifier number is CA17-00071. Written informed consent was obtained from all participants. The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Independent Ethics Committee/Institutional Review Board of each participating center as shown in Supplementary Table 1 (available online).
Measurement of α-galactosidase A activity
In female patients, α-Gal A enzyme activity was measured using whole blood. Leukocytes were isolated [7], and α-Gal A enzyme activity was measured with a fluorometer (1420 Multi-label Counter; PerkinElmer). The substrate and inhibitor used were 4-methylumbelliferyl-α-D-galactopyranoside (Sigma-Aldrich) and N-acetylgalactosamine (Sigma-Aldrich), respectively. Enzymatic activity was evaluated using the calibration curve of 4-methylumbelliferone (Sigma-Aldrich) and expressed as nmol/hr/mg protein. In male patients, α-Gal A enzyme activity was measured from dried venous blood spots on a Whatman 903 filter paper (GE Healthcare Life Sciences). The separation and detection of α-Gal A was performed using a high-performance liquid chromatography system (HPLC system; Agilent 1200 series, Agilent) and a flow injection analysis-tandem mass spectrometry system (FIA-MS/MS, API 4000, SCIEX) operated in a multiple reaction monitoring mode using NeoLSDTM MSMS KIT (PerkinElmer). The enzyme activity was calculated and expressed as µmol/hr/L.
Measurement of plasma lyso-GL3
Plasma lyso-GL3 was measured by liquid chromatography-tandem mass spectrometry. HPLC was performed with Agilent 1200 and Unison UK-C18 column (2.0 Ã 50 mm, 3 ÎŒm; Imtakt), followed by an MS/MS analysis (API 4000).
Genetic analysis
Genetic analysis was performed with the Sanger sequencing method. Seven exons and flanking intron sequences of the GLA gene were amplified via polymerase chain reaction using ProFlex PCR System (Applied Biosystems) and sequenced using a 3500Dx Genetic Analyzer (Applied Biosystems) according to the manufacturerâs instruction. Analysis of the sequences was done with SeqScape v2.7 (Applied Biosystems). The references used for the identification of mutation and the determination of pathogenicity are as follows: the Human Genome Mutation Database (www.hgmd.org), the Fabry Database of Fabry disease mutations (http://fabry-database.org/), the American College of Medical Genetics standards and guidelines for the interpretation of sequence variants [8], and the International Fabry disease Genotype-Phenotype Database (dbFGP) (www.dbfgp.org).
Results
Baseline characteristics
A total of 902 patients were screened, of which five were excluded since they did not give consent to an amended informed consent form, thus 897 patients were eligible and included in this analysis. Of the total 897 patients, 405 (45.2%) were males and 492 (54.8%) were females and 279 of 897 patients (31.1%) were on dialysis; of the 279 patients who were on dialysis, 196 patients (70.3%) were on hemodialysis, and the remaining 83 (29.7%) were on peritoneal dialysis. The proportion of patients who were on dialysis in the male and female groups was 34.1% and 28.7%, respectively. Of the 42 patients with kidney transplants, 36 patients were on dialysis at the time of screening. And 50 of 897 patients (5.6%, 25 treated on dialysis) had kidney biopsy findings that were not informative to conclude a specific disease. The mean age of the male and female patients was 46.8 ± 9.5 years and 55.5 ± 13.5 years, respectively; almost all of the patients were Korean (888 of 897, 99.0%). The demographic and baseline characteristics are shown in Table 1. Values of estimated glomerular filtration rate (eGFR) were reported in 478 of 618 (77.3%) of nondialysis patients. Blood pressure and body mass index were reported in 856 of 897 (95.4%) and 849 of 897 (94.6%) of all patients, respectively.
FD-related symptoms were reported in patients at only two sites; therefore, these results needed to be interpreted with caution. The most frequently reported symptom was cardiac dysfunction (21 of 879, 2.3%), followed by gastrointestinal and ocular symptoms (12 of 897, 1.3% each). Dermatologic, neurologic, and ENT symptoms, were respectively reported in seven patients (0.8%) (data not shown).
Screening tests: α-galactosidase A enzyme activity and plasma lyso-GL3 concentration
In total, α-Gal A enzyme activity was measured in 99.3% (891 of 897) of the patients using either DBS (for male patients) or whole blood (for female patients). All 405 male patients underwent a DBS test, with the results reported as normal or abnormal without enzyme activity measurement. Four male patients, all on dialysis, showed an abnormal DBS test (â€2.35 ÎŒmol/hr/L). Whole blood α-Gal A enzyme activity was measured in 98.7% of female patients (486 of 492). Four female patients, of whom three were on dialysis, showed a decreased enzyme activity, with a mean value of 32.3 ± 1.64 nmol/hr/mg protein. The mean value of normal results was 67.9 ± 19.15 nmol/hr/mg protein (range, 36.3â139.8 nmol/hr/mg protein).
The plasma lyso-GL3 concentration was measured in all 897 patients. Two patients, who were not on dialysis and with normal enzyme results, showed an increased plasma lyso-GL3 concentration of 3.57 and 5.66 ng/mL, compared to the mean value of normal concentration which was 1.001 ± 0.045 ng/mL. However, eight patients with decreased enzyme activity, four in DBS and four in whole blood, exhibited normal plasma lyso-GL3 concentrations, as shown in Table 2. Therefore, 10 patients were eligible for a GLA gene test to confirm FD. All these patients were of Korean ethnicity, and none of them were kidney transplant recipients. The clinical and laboratory data of patients with abnormal screening results are summarized in Table 2. The mean age of the patients eligible for a gene test was 44.0 ± 8.9 years, and seven patients were on dialysis. Only nine patients were subjected to a gene test, because one patient did not submit the informed consent for gene analysis. Among those nine patients, two female patients with an elevated lyso-GL3 concentration had a kidney biopsy done, which will be discussed later.
GLA gene analysis and a case presentation
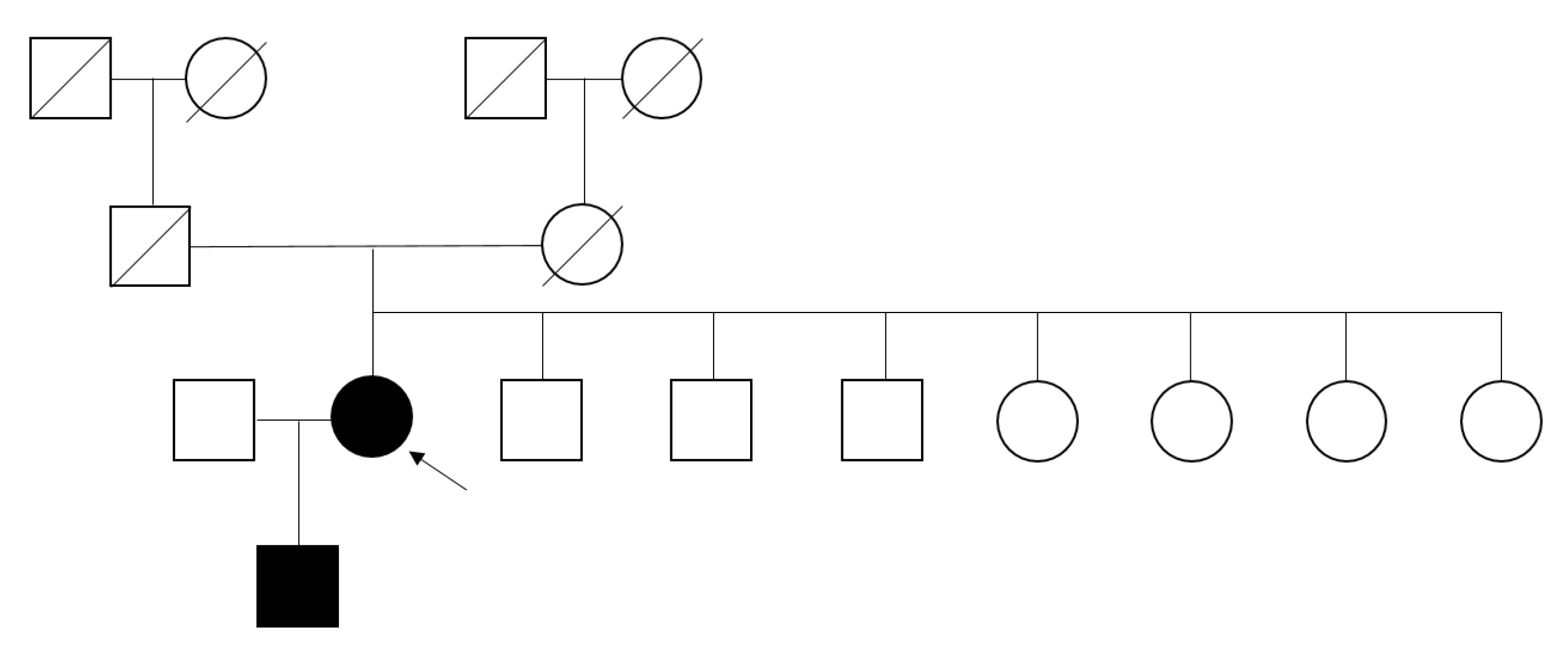
Mutation analysis of the GLA gene was performed in 1% (9 of 897) of the total study population, and FD was diagnosed in one patient, resulting in a prevalence of 0.11% (1 of 897). Gene analysis detected a missense mutation in exon 5 of the GLA gene with the c.778G>C variant, resulting in an amino acid change of p.Gly260Arg, which was reported to be pathogenic and related to a classic phenotype at dbFGP (Fig. 2). This mutation was found in a 50-year-old female patient, not on dialysis, with an increased plasma lyso-GL3 concentration of 5.66 ng/mL and normal enzyme activity of 51.6 nmol/hr/mg protein. Her eGFR was 147.5 mL/min/1.73 m2, and she had high PCR results of 980 and 1,200 mg/g. She did not have an underlying disease, such as hypertension or diabetes mellitus (DM), but suffered from unexplained acroparesthesia. Her electrocardiogram (ECG) showed T-wave inversion in anterolateral and inferior leads (Fig. 3) without abnormal findings on the echocardiogram (left ventricular mass index of 82.9 g/m2, relative wall thickness of 0.39, and septal wall thickness of 10 mm), and electron microscopy of her kidney biopsy demonstrated myeloid bodies in podocytes, glomerular and mesangial cells, and vessels. The patient underwent ERT.
Family screening
Family screening was performed, which revealed that her 26-year-old son had the same mutation (Fig. 4). He presented acroparesthesia and hypohidrosis. This c.778G>C (p.G260R) hemizygote patient showed abnormal α-Gal A enzyme activity on a DBS test (0.15 µmol/hr/L) as well as a remarkably increased plasma lyso-GL3 concentration of 116 ng/mL and an increased plasma GL-3 concentration of 13.6 µg/mL (normal, 3.9â9.9 µg/mL). He had albuminuria (ACR of 40.68 mg/g) at the time of screening, which elevated to 87.0 mg/g after 2 years. However, the patient rejected further evaluation regarding FD.
Discussion
During the study period, 897 patients were screened. One patient was identified with FD, exhibiting a 0.11% prevalence of FD in the CKD population in South Korea. To the best of our knowledge, three screening studies for the prevalence of FD have been conducted in Korea so far [2â4]. One of them screened 480 patients on dialysis using serum GL-3 but did not observe any patient with FD. The other two studies included patients with hypertrophic cardiomyopathy and male patients with left ventricular hypertrophy. Regarding Fabry nephropathy, most screening studies have been performed outside of Korea, and most of them included male patients or patients who were on dialysis. Doheny et al. [9] reanalyzed the prevalence results of previous screening studies conducted between 1995 and 2017 with regard to pathogenicity of GLA gene mutation. The results reported that the prevalence of FD was 0.21% and 0.15% for male and female patients, respectively, on hemodialysis, and 0.25% and 0% for male and female patients, respectively, who received renal transplants [9]. Also, a screening study that involved 1,453 CKD patients (not on dialysis) was performed in Turkey, which reported the prevalence of FD as 0.2% (0.4% in male, 0% in female) [10]. Although male and female patients were recruited to the present study in a nonselective way including both CKD patients with and without dialysis, the prevalence rate was similar to previous reports, albeit slightly low [9,10]. Difference in estimated prevalence rates could be attributed to the selection of study population, screening methods used, and the choice of cutoff values [11].
Renal involvement in FD often begins with hyperfiltration followed by proteinuria and a decline in the glomerular filtration rate, ultimately leading to end-stage renal disease and a decreased life expectancy [5,6]. The age at which ERT is initiated is an important factor related to progressive loss of kidney function; additionally, screening is essential for early diagnosis [12â14]. The European Renal Best Practice Group recommends screening for FD in male patients below 50 years old and in female patients of any age with unexplained CKD [1]. However, after the introduction of dialysis and renal transplantation, the average age at death was reported to be 50 to 57 years in male patients and 64 to 72 years in female patients [15]. Therefore, the present study recruited male CKD patients aged â€60 years to encompass possible undiagnosed FD patients. Renal manifestations are nonspecific, and the progression of CKD in Fabry nephropathy resembles that of diabetic nephropathy [11]. Hypertension and DM, the most common causes of CKD, are also frequent in patients with FD [16]. Multiple symptoms of DM may overlap with those of FD, thus making the diagnosis more difficult. Moreover, FD can be diagnosed in patients with diabetic and hypertensive nephropathy. Patients with dual pathology are observed, although it is rare [10,17]. The present study did not assess the prevalence of hypertension and DM, and patients with known diabetic nephropathy were excluded as decided by the responsible physician. This might have affected the prevalence rate of FD in the CKD population observed in this study.
This study identified only one female patient with FD. The low prevalence might be caused by the presence of 55% female patients in the study population. Because the values of α-Gal A enzyme activity in heterozygous female patients are divergent, and up to one-third showed an enzyme activity within the normal range due to a random X-chromosome inactivation [10,18]. Similarly, clinical symptoms in female patients are more diverse, from asymptomatic to severe, often at a later age in comparison to the symptoms observed in male patients, which are related to residual enzyme activity. Therefore, GLA gene mutation analysis is suggested as a primary screening tool in female patients, whereas enzyme activity measurement is recommended in male patients [1]. The latter method has been primarily used in the majority of screening studies partly due to its convenience and cost-effectiveness, in addition to avoiding unnecessary gene analysis [10]; nevertheless, gene mutation test is essential to diagnose FD, even though it is not always an ideal approach. Nearly 1,000 GLA gene mutations have been reported, and there are many gene variants of unknown significance (GVUS) [19]. For individuals with a GVUS of GLA gene, enzyme activity, plasma lyso-GL3, and/or evidence of GL-3 accumulation in an affected organ need to be studied, as they can help in the diagnosis of FD. Earlier studies have indicated the availability of reliable biomarkers for FD that can identify candidates for gene analysis more easily [20â25]. This study measured the α-Gal A enzyme activity using DBS in male patients, which is convenient and less expensive, and has a nearly 100% negative predictive value in male patients [1,26,27]. However, it used whole blood (leukocyte) in female patients, the gold standard for measuring the enzyme activity, because of a higher false-negative value with using DBS compared to using leukocytes in females [18]. The plasma lyso-GL3 was also measured in all patients, regardless of the results of enzyme activity.
Interestingly, no patient showed concurrent abnormal results in terms of both α-Gal A enzyme activity and plasma lyso-GL3. However, FD was diagnosed only in a patient with elevated plasma lyso-GL3. This result is in line with a previous study [20] where the investigators measured plasma lyso-GL3 concentration and plasma α-Gal A enzyme activity for screening of FD and identified 13 patients with FD (seven male and six female patients) out of 2,360 screened patients. Likewise, all patients with FD were detected in the group with elevated plasma lyso-GL3 [20]. Of note, a female patient with FD with elevated plasma lyso-GL3 displayed a normal α-Gal A enzyme activity. Nonpathogenic mutations or GVUS were found in patients with a low α-Gal A enzyme activity and normal plasma lyso-GL3. Although not all patients were genotyped and there were differences in the cutoff values and the method of α-Gal A enzyme measured [20], the results were similar to those obtained in the present study. In this study, a nucleotide substitution at c.-10C>T was also identified in two female patients with a low α-Gal A enzyme activity and normal plasma lyso-GL3, which is considered as a benign germline variant (https://www.ncbi.nlm.nih.gov/clinvar/RCV000335296/). This mutation was in exon 1 of the 5â untranslated region upstream of the coding sequence and this region is often involved in the regulation of protein translation. This variant was classified as benign as per American College of Medical Genetics and Genomics guidelines. However, to determine its clinical significance, additional test like complex intronic haplotype (c.-10C>T, c.369+990C>A, c.370-81_370-77delCAGCC, c.640-16A>G, c.1000-22C>T) needs to be done. Routine GLA gene analysis which examines exons and flanking introns cannot detect a deep intronic mutation, although pseudoexon-activating mutations are often located deep in the introns [28,29].
In addition, the study by Maruyama et al. [20] evaluated patients screened positive for lyso-GL3 and having normal α-Gal A enzyme activity and a negative result in the gene test, and lamellar bodies were found in kidney or endomyocardial biopsies. Similarly, in our study, kidney biopsy from a female patient with elevated plasma lyso-GL3 (3.57 ng/mL) and negative gene test (Table 2) exhibited signs of FD with a large amount of laminated electron-dense bodies observed in the podocyte cytoplasm. She had eGFR of 81.8 mL/min/1.73 m2 and proteinuria (PCR) of 990 and 830 mg/g. Her ECG showed a right bundle branch block and blood pressure was 109/75 mmHg at the time of inclusion. She did not present FD-related symptoms and there was no information about her medication history. However, it is suggested that this patient needs to be closely monitored for disease progression and the possible diagnosis of FD in due course of time.
Plasma lyso-GL3 analysis has been shown to be a more sensitive and specific test than α-Gal A enzyme activity assays for the diagnosis of FD, especially in heterozygous female patients [20â24]. Plasma lyso-GL3 is also suggested as a useful biomarker for therapeutic monitoring [30]. However, it has been proved that the values of lyso-GL3 increase with age in female patients, and the normal levels of lyso-GL3 cannot confirm the absence of FD. Recently, Baydakova et al. [25] suggested the α-Gal A/lyso-GL3 ratio as a novel biochemical criterion to increase sensitivity for diagnosis of FD in female patients. Although evaluating the role of plasma lyso-GL3 is out of the scope of this study, the results support the view that the measurement of both α-Gal A enzyme activity and lyso-GL3 helps to improve the screening ability for the diagnosis of FD.
There are a few limitations in the present study. First, the sample size is moderate, and the proportion of RRT in screened patients is not representative of CKD patients in South Korea. Second, not all participants have undergone gene analysis, which leaves the possibility of missing unrecognized FD patients. Third, α-Gal A enzyme activity or plasma lyso-GL3 can remain within normal range in female patients with FD. However, in this study, a dual-screening method was used involving both α-Gal A enzyme activity and lyso-GL3, which makes it less likely to overlook adult female patients with FD. Fourth, as only two institutions reported FD-related symptoms, there are limitations to interpreting the data. Fifth, since patients considered to have typical diabetic nephropathy were excluded on the discretion of the responsible physician, there is a possibility that patients who have both diabetes and FD might be excluded.
In the present study, the prevalence of FD in CKD patients via screening tests was low (0.11%); however, identification of one patient provides an opportunity for early treatment to patientâs relatives. We also determined that both α-Gal A enzyme activity and plasma lyso-GL3 concentration tests are needed to identify patients who are eligible for gene analysis. Further studies are required to examine the relationship between plasma lyso-GL3 concentration and the GLA gene mutation. In patients with CKD of unknown etiology, clinicians should consider FD as a differential diagnosis.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement table 1
Supplement table 1 Print
Print






")









