



Introduction
Diabetic kidney disease (DKD) is a major complication of diabetes mellitus that can lead to end-stage kidney disease (ESKD), cardiovascular disease, and high mortality [1].
Conventionally, chronic kidney disease (CKD) caused by diabetes mellitus is diagnosed as diabetic nephropathy. The typical clinical manifestation starts with glomerular hypertrophy and hyperfiltration, followed by microalbuminuria that progresses to macroalbuminuria [2]. Subsequently, kidney function declines gradually, leading to ESKD. DKD is characterized histologically by mesangial matrix expansion, thickening of the glomerular basement membrane, and the presence of nodular lesions [3]. However, in recent years, it has become apparent that some patients with diabetes mellitus present with a low glomerular filtration rate (GFR) without albuminuria [4]. The redefined term ‘DKD’ includes not only the conventional proteinuric nephropathy of diabetes mellitus but also nephropathy without overt albuminuria. In this context, it is assumed that the diabetic population is aging, and the prevalence of comorbidities such as hypertension and dyslipidemia, which increase nephrosclerosis, is growing. Until recently, managing blood glucose and blood pressure with renin-angiotensin system (RAS) inhibitors (such as angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers [ARBs]), lipid control, and weight management have been the standard therapeutic strategy for DKD. However, even that comprehensive treatment cannot entirely arrest the progression of DKD to ESKD (residual risk), and the prevalence of DKD among diabetic patients has not changed significantly during the past decade [4].
The approach to glycemic control in patients with DKD has started to change. Dipeptidyl peptidase-4 inhibitors and glucagon-like peptide 1 (GLP-1) receptor agonists, which are both incretin-related drugs, appeared as novel hypoglycemic agents in the 2000s, and they were followed by sodium-glucose cotransporter 2 (SGLT2) inhibitors. Recent clinical trials have revealed that some of those agents not only lower blood glucose levels but also protect renal function.
Additional novel therapeutic agents have been developed and examined in animal models and clinical trials. Here, we review the pathophysiology and recently revealed effects of existing drugs and outline prospective therapeutic candidates for DKD.
Pathophysiology of diabetic kidney disease
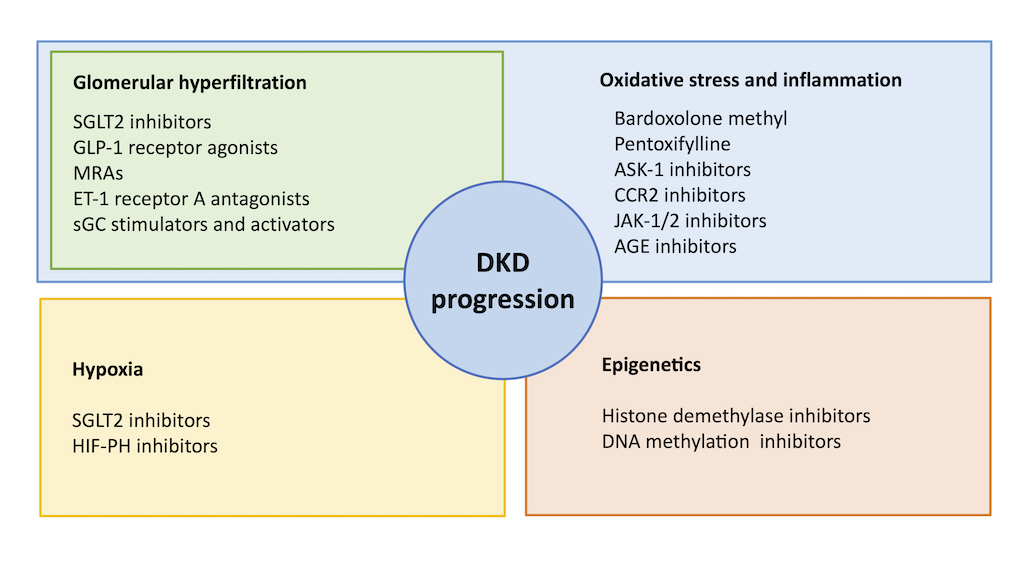
The pathophysiology of DKD is multifactorial, complex, and not fully understood. Many new agents in use and development focus on specific mechanisms and relevant molecular pathways, as shown in Fig. 1. In this section, we review the pathophysiology of DKD by dividing it into several factors.
Glomerular hypertension and hyperfiltration
In the early phase, hyperglycemia causes glomerular hypertension and hyperfiltration of the kidneys. Glomerular hyperfiltration is suspected to be a risk factor for albuminuria and future decreases in the estimated GFR (eGFR) [5]. Increased glomerular capillary pressure heightens the tensile stress on the capillary wall, and the consequent increase in ultrafiltrate flow into Bowman’s space elevates the shear stress on the podocyte foot processes and body surface. These mechanical stresses induce an increase in the glomerular basement membrane length, podocyte hypertrophy, and eventually podocyte loss and segmental sclerosis. Hyperfiltration and the consequent increase in proximal tubular flow increase the delivery and reabsorption of small- and large-molecular-weight solute, with ensuing tubulointerstitial inflammation, hypoxia, and fibrosis [6].
The mechanisms of hyperfiltration can be explained in three ways. First, a kidney with DKD becomes large due to an expansion in nephron size caused by various cytokines and growth factors in response to hyperglycemia. The increased kidney size and filtration surface area per glomerulus are thus related to hyperfiltration. Second, the imbalance of vasoactive factors controls efferent and afferent arteriolar constriction and dilation. For example, angiotensin II and endothelin-1 (ET-1) mediate glomerular hyperfiltration by increasing efferent arteriolar resistance [5]. Third, glomerular hyperfiltration is caused by dysregulated tubuloglomerular feedback. Under hyperglycemic conditions, excessive glucose is filtered at the glomeruli and reabsorbed via SGLTs in the proximal tubules. Sodium is also reabsorbed with glucose at SGLTs, so the amount of sodium that reaches the distal tubular macula densa decreases. To keep the amount of sodium constant, a feedback mechanism is set in motion such that afferent arterioles expand while efferent arterioles contract relatively, resulting in glomerular hypertension and hyperfiltration. These three mechanisms are thought to be the major causes of hyperfiltration.
Oxidative stress and inflammation
Oxidative stress and inflammation also play central roles in the progression of DKD. Hyperglycemia activates several intracellular metabolic pathways: the polyol pathway, the advanced glycation end-product (AGE) formation, the activation of protein kinase C (PKC) isoforms, and the hexosamine pathway, all of which lead to the generation of reactive oxygen species (ROS) [7].
The PKC pathway also stimulates nuclear factor kappa B (NF-κB) and releases tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) [8]. In addition, PKC activation has various pathogenic consequences by affecting the expression of endothelial nitric oxide synthetase (eNOS), ET-1, vascular endothelial growth factor, transforming growth factor-β (TGF-β), and plasminogen activator inhibitor-1 [7].
Chronic hyperglycemia increases the expression and activity of various nicotinamide adenine dinucleotide phosphate oxidase (NOX) isoforms and generates high levels of ROS. Among the NOX isoforms, NOX4- and NOX5-derived ROS have been suggested to mediate glomerular injury and podocytopathy, leading to albuminuria in DKD [9]. Increased angiotensin II levels induce oxidative stress through NOX activation [10]. PKC-α and PKC-β also play important roles in diabetic renal pathology via NOX activation [9].
Mitochondrial dysfunction is another source of ROS. Under hyperglycemic conditions, more glucose-derived pyruvate is oxidized in the mitochondria, which pushes more electron donors into the electron transport chain and leads to electron leakage, resulting in the overproduction of superoxide [11].
Excessive ROS production leads to renal fibrosis and inflammation and induces tissue damage through different mechanisms, including lipid peroxidation, DNA damage, protein modification, and mitochondrial dysfunction [12].
Hyperglycemia induces AGEs, which are the final products of non-enzymatic glycation and accumulate in CKD patients because of reduced clearance [13]. AGEs activate various pathways, such as the phosphatidylinositol 3-kinase, Janus kinases (JAK), and mitogen-activated protein kinase (MAPK) pathways, by activating AGE receptors (RAGE). RAGE activation also increases oxidative stress and activates NF-κB [14].
Many more pathways interact and are related to oxidative stress and inflammation in ways that affect DKD progression, and new medications targeting those mechanisms have been developed (Table 1).
Hypoxia
Renal hypoxia is an important factor in DKD progression and a common final pathway to ESKD, regardless of the cause of CKD. However, this condition is exacerbated in DKD due to the mismatch between oxygen demand and supply. Oxygen demand increases due to glomerular hyperfiltration and elevated activity of SGLT2 under hyperglycemic conditions. Oxidative stress also induces renal fibrosis and contributes to renal hypoxia by decreasing oxygen delivery. Oxidative stress increases intracellular oxygen consumption, resulting in a demand/supply mismatch for oxygen and progressive hypoxia.
Epigenetics
Epigenetics is a regulatory mechanism for controlling gene expression without changing DNA sequences. This mechanism includes chromatin histone modifications, DNA methylation, and noncoding RNAs. Metabolic memory is a phenomenon by which DKD-related gene and phenotype expression caused by prior hyperglycemia continue even after blood sugar levels become normal in response to appropriate treatment. Epigenetic mechanisms contribute to metabolic memory by inducing the persistent expression of pathogenic genes, even after normoglycemia has been established [15].
Renoprotective effects of sodium-glucose cotransporter 2 inhibitors, glucagon-like peptide 1 receptor agonists, and mineralocorticoid receptor antagonists
To date, RAS inhibitors have been at the center of DKD treatment because they reduce albuminuria and slow the decline in eGFR. RAS inhibitors function mainly by reducing intraglomerular pressure and hyperfiltration and repressing oxidative stress. Their effects in DKD patients have been demonstrated in several randomized clinical trials, such as the collaborative study, RENAAL (Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan), and IDNT (Irbesartan Diabetic Nephropathy Trial) [16–18]. However, RAS inhibitors cannot completely halt DKD progression. Intensive RAS inhibition was examined in combination with ACE inhibitors and ARBs in the ONTARGET (Ongoing Telmisartan Alone and in Combination with Ramipril Global End-point Trial), but the combination therapy significantly increased the worsening of kidney function and DKD-related mortality [19]. In the VA NEPHRON-D (Veterans Affairs Nephropathy in Diabetes) trial, another combination therapy of ACE inhibitors and ARBs also showed increased adverse events, hyperkalemia, and acute kidney injury [20].
Novel treatments have been explored for a long time, and the renoprotective effects of existing drugs have recently attracted attention. The effectiveness of SGLT2 inhibitors has been demonstrated, and SGLT2 inhibitors have been approved for CKD treatment. The effects of GLP-1 receptor agonists and mineralocorticoid receptor antagonists (MRAs) have also been explored, and we review those results next.
Sodium-glucose cotransporter 2 inhibitors
SGLT2 is a cotransporter that transports sodium and glucose using a sodium gradient established by the Na+/K+ ATPase pump. SGLT2 inhibitors are hypoglycemic agents that suppress glucose reabsorption in the proximal tubules and stimulate glucose excretion in the urine. SGLT2 inhibitors also suppress the reabsorption of sodium along with glucose, which corrects the hyperglycemia-induced disorder of tubuloglomerular feedback and ameliorates glomerular hyperfiltration in DKD. SGLT2 inhibitors reduce mitochondrial damage, NOX4 expression, and subsequent oxidative stress [21,22]. In the kidneys of diabetic mice, SGLT2 inhibitors normalized glucose metabolism by eliminating accumulated tricarboxylic acid cycle intermediates, which suppressed the increase in oxidative stress [23]. SGLT2 inhibitors also improve renal hypoxia. Oxygen consumption in the kidney is attributed to Na+/K+ ATPase, and SGLT2 inhibitors can suppress the increase in Na+/K+ ATPase activity caused by SGLT2 [24]. SGLT2 inhibitors increase the production of ketone bodies, which can produce more ATP than glucose or fatty acids with a small amount of oxygen. SGLT2 inhibitors were reported to improve anemia [25] and ketone bodies can also correct the hyperactivation of the mechanistic target of rapamycin complex 1 [26], which produces further renoprotective effects.
The renoprotective effects of SGLT2 inhibitors in established DKD were demonstrated in the CREDENCE (Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation) trial of canagliflozin [27]. Recently, the DAPA-CKD (Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease) trial, a phase 3 clinical trial of dapagliflozin, showed that in CKD patients with or without type 2 diabetes mellitus, dapagliflozin significantly lowered the composite renal risk of a sustained decline in eGFR of at least 50%, ESKD, and death from renal or cardiovascular causes compared with those who received placebo [28]. Based on those results, the U.S. Food and Drug Administration approved dapagliflozin for CKD treatment. This is the first SGLT2 inhibitor to be approved to treat CKD. The EMPA-KIDNEY (the Study of Heart and Kidney Protection With Empagliflozin) trial, a phase 3 study of empagliflozin, is in progress to investigate the effects of empagliflozin on kidney disease progression and cardiovascular death in CKD patients. It is encouraging that SGLT2, originally developed as a hypoglycemic agent, now provides an additional choice for CKD treatment.
Glucagon-like peptide 1 receptor agonists
GLP-1 is an incretin hormone that stimulates insulin secretion and suppresses glucagon secretion in response to food intake. GLP-1 receptor agonists have been used as hypoglycemic agents, and their potential to offer renal protection is of great interest.
GLP-1 receptor agonists inhibit Na+/H+ exchanger isoform 3 and reduce proximal sodium reabsorption [29]. As with SGLT2 inhibitors, GLP-1 receptor agonists relieve hyperfiltration by correcting abnormal tubuloglomerular feedback. GLP-1 receptor agonists have also been reported to have antioxidative and anti-inflammatory effects in animal models. In a rat model of streptozotocin-induced type 1 diabetes mellitus, activation of the GLP-1 receptor with exendin-4 ameliorated albuminuria, glomerular hyperfiltration, glomerular hypertrophy, and mesangial matrix expansion without changing blood pressure or body weight. Exendin-4 also prevented macrophage infiltration and decreased protein levels of intercellular adhesion molecule-1 (ICAM1) and type IV collagen, as well as decreasing oxidative stress and NF-κB activation in kidney tissue [30].
In the LEADER (Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results) and SUSTAIN-6 (Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semaglutide in Subjects with Type 2 Diabetes) trials, liraglutide and semaglutide, respectively, were shown to reduce the frequency of cardiovascular events in type 2 diabetes mellitus patients with high cardiovascular risk [31,32]. A significant reduction in negative renal outcomes was observed in both trials, but they were mainly due to a decrease in new-onset persistent macroalbuminuria; the effects of those agents on hard renal endpoints remain elusive [32,33]. The AWARD-7 (Dulaglutide Versus Insulin Glargine in Patients With Type 2 Diabetes and Moderate or Severe CKD) trial showed that 52 weeks of once-weekly dulaglutide treatment significantly reduced eGFR decline compared with insulin glargine in patients with type 2 diabetes and moderate-to-severe CKD [34]. The AMPLITUDE-O (Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes) trial demonstrated that weekly subcutaneous injections of efpeglenatide, an exendin-based GLP-1 receptor agonist, lowered the risk of cardiovascular events in patients with type 2 diabetes who had either a history of cardiovascular disease or current kidney disease plus at least one other cardiovascular risk factor [35]. Additionally, efpeglenatide led to a 32% lower risk of composite renal outcomes (a decrease in kidney function or macroalbuminuria) than placebo in that trial. Currently, the FLOW (Effect of Semaglutide Versus Placebo on the Progression of Renal Impairment in Subjects With Type 2 Diabetes and Chronic Kidney Disease) trial is progressing to examine the 5-year use of semaglutide, which might clarify the hard renal outcomes of long-term treatment.
Mineralocorticoid receptor antagonists
MRAs, such as spironolactone and eplerenone, are widely used as antihypertensive drugs and work by suppressing aldosterone activity. Preclinical data showed that aldosterone increases mineralocorticoid receptor-dependent NOX activity and ROS, induces a proinflammatory state, stimulates fibrosis, regulates vascular tone, and affects renal hemodynamics [36]. Addition of spironolactone on the RAS blockade has been shown to decrease albuminuria in DKD [37]. However, because of the risk of hyperkalemia, its use is limited in CKD patients.
Recently, nonsteroidal MRAs, esaxerenone, finerenone, and apararenone, have been developed. They have higher selectivity and affinity to mineralocorticoid receptors than steroidal MRAs such as spironolactone and eplerenone, which might lower the risk of hyperkalemia.
A phase 3 clinical trial of esaxerenone, ESAX-DN (Esaxerenone [CS-3150] in Patients with Type 2 Diabetes and Microalbuminuria) trial, demonstrated that adding esaxerenone to existing RAS inhibitor therapy in patients with type 2 diabetes and microalbuminuria increased the likelihood that albuminuria would return to normal levels and reduced the progression of albuminuria to higher levels [38]. A phase 3 trial of finerenone, FIDELIO-DKD (Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease) trial, confirmed that in patients with CKD and type 2 diabetes, treatment with finerenone lowered the risk of CKD progression, including kidney failure, a sustained decrease of ≥40% in the eGFR from baseline, and death from renal causes, compared with placebo [39]. Hyperkalemia was observed as an adverse event, but its frequency was markedly lower than in trials of a dual RAS blockade. We need to pay attention to hyperkalemia, but nonsteroidal MRAs could be an additional option for DKD treatment.
Finerenone also reduced the risk of cardiovascular events, and a reduced risk of death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke, and hospitalization for heart failure was observed in a wide range of CKD patients with type 2 diabetes (FIGARO-DKD [Finerenone in Reducing Cardiovascular Mortality and Morbidity in Diabetic Kidney Disease] trial) [40]. In the FIGARO-DKD trial, there was no significant difference between the finerenone and placebo groups in the incidence of the first secondary composite renal outcome of kidney failure, a sustained decrease from baseline of at least 40% in the eGFR, or death from renal causes. The mean eGFR of enrolled patients was higher in the FIGARO-DKD trial than the FIDELIO-DKD trial, so more time would be needed to achieve the secondary outcome in the FIGARO-DKD trial, which might explain why it could not confirm a significant difference in the incidence of the first secondary composite renal outcome. Nonetheless, the effects of finerenone treatment on the kidney were similar in the FIGARO-DKD and FIDELIO-DKD trials.
New molecular targets and treatment options
Many new prospective therapeutic agents have been developed, and some of them have been subjected to clinical trials.
Bardoxolone methyl
Treatment of DKD has focused on suppressing the decline in kidney function. Bardoxolone methyl was the first agent to show an improvement in GFR in clinical trials, which has attracted attention.
Bardoxolone methyl is a synthetic triterpenoid that activates the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which has anti-inflammatory and antioxidant effects. Kelch-like ECH-associated protein 1 (Keap1) forms an ubiquitin E3 ligase complex with Cullin3 and polyubiquitinates Nrf2 in the cytoplasm. Under normal conditions, Nrf2 is constantly degraded. However, under oxidative stress, the reactive cysteine residues of Keap1 are directly modified, and ubiquitin E3 ligase activity decreases, which leads to the stabilization of Nrf2. Nrf2 then translocates into the nucleus and binds to antioxidant response elements with small musculoaponeurotic fibrosarcoma and induces the expression of its target genes [41]. Nrf2 regulates the expression of more than 250 genes, including those encoding antioxidant and phase 2 detoxifying enzymes and related proteins, such as catalase, superoxide dismutase, uridine 5'-diphospho-glucuronosyltransferase, NAD(P)H: quinone oxidoreductase-1, heme oxygenase-1, glutamate cysteine ligase, glutathione S-transferase, glutathione peroxidase, and thioredoxin [42]. Bardoxolone methyl induces conformational changes in Keap1 that enable Nrf2 to translocate into the nucleus and exhibit its anti-inflammatory and antioxidant effects. It was reported that Nrf2 suppressed the macrophage inflammatory response by blocking the transcription of proinflammatory cytokines [43]. Bardoxolone methyl also blocks the NF-κB pathway by suppressing the inhibitor of the NF-κB kinase subunit β [44].
Bardoxolone methyl was first developed as an antitumor agent. However, in a clinical trial for malignancy, it was found to increase eGFR, so the study of it expanded to the renal area. Bardoxolone methyl is related to anti-inflammatory and antioxidant factors; therefore, its efficacy in DKD was considered because both factors affect DKD progression. In the phase 2 BEAM (52-Week Bardoxolone Methyl Treatment: Renal Function in CKD/Type 2 Diabetes) clinical trial, bardoxolone methyl significantly increased the eGFR of patients with type 2 diabetes and CKD whose eGFR was 20 to 45 mL per minute per 1.73 m2 of body surface area [45]. However, the phase 3 BEACON (Bardoxolone Methyl Evaluation in Patients with CKD and Type 2 Diabetes Mellitus: the Occurrence of Renal Events) clinical trial enrolling patients with type 2 diabetes and stage 4 CKD was terminated prematurely because of the high rate of cardiovascular events in patients receiving bardoxolone methyl [46]. Subsequent analyses revealed that elevated baseline B-type natriuretic peptide and prior hospitalization for heart failure were risk factors for cardiovascular events associated with bardoxolone methyl treatment [47]. In Japan, another phase 2 clinical trial, the TSUBAKI (The Phase 2 Study of Bardoxolone Methyl in Patients with CKD and Type 2 Diabetes) trial, was initiated and enrolled only type 2 diabetes and CKD stage 3–4 patients without previous cardiovascular risk factors [48]. The bardoxolone methyl group in that trial saw significant increases in GFR measured by inulin clearance without any cardiovascular deaths or cases of heart failure. That study suggested that bardoxolone methyl might be a safe and effective medication for DKD patients free of the relevant risk factors. To examine its safety and efficacy in the longer term, a phase 3 trial, the AYAME (A Phase 3 Study of Bardoxolone Methyl in Patients With Diabetic Kidney Disease) trial, is currently in progress.
Some clinical trials have reported that bardoxolone methyl increases albuminuria [49], possibly as a result of the increase in GFR [50]. Decreasing the reabsorption of albumin at the proximal tubules relieves inflammation of the proximal tubules, which might be related to the improvement in GFR [49,50]. However, it is still not well understood whether the increase in GFR and albuminuria protectively affect DKD over a long period. The AYAME trial is expected to clarify these points.
Endothelin-1 receptor A antagonists
ET-1 is a vasoconstrictor peptide that is mainly secreted from endothelial cells and acts through two types of receptors: ET-A and ET-B receptors. ET-1 was reported to constrict afferent arterioles more strongly than efferent arterioles via ET-A receptors in a mouse model [51]. ET-A receptor activation also contributes to pro-apoptosis pathways, leading to the loss of podocytes, albuminuria, and the destruction of glomerular capillaries in vivo [52]. ET-1 damages the vasculature, induces fibrosis, and promotes inflammatory cell infiltration, and in mesangial cells, ET-1 causes proliferation and extracellular matrix accumulation [53]. Therefore, it is hypothesized that ET-A receptor antagonists might be therapeutic agents for DKD. However, the ASCEND (A Randomised, Double Blind, Placebo Controlled, Parallel Group Study to Assess the Effect ofthe Endothelin Receptor Antagonist Avosentan on Time to Doubling of Serum Creatinine, End Stage Renal Disease or Death in Patients With Type 2 Diabetes Mellitus and Diabetic Nephropathy) phase 3 clinical trial was terminated prematurely because of an increased incidence of cardiovascular events in the avosentan group, mainly congestive heart failure and fluid overload [54]. A high dose of avosentan might be inadequately selective for the ET-A receptor and block ET-B receptors which inhibit sodium reabsorption at the proximal tubules and collecting ducts. Based on that result, an antagonist with higher efficacy and selectivity for the ET-A receptor, atrasentan, was examined in the short term and found to reduce albuminuria without causing significant fluid retention [55]. After that, the SONAR phase 3 clinical trial of atrasentan was initiated [56]. In the SONAR (Study Of Diabetic Nephropathy With Atrasentan) trial, responders to atrasentan who saw reduced albuminuria without fluid retention during the initial 6-week open-label period were enrolled and randomized. During a median follow-up of 2.2 years, atrasentan significantly lowered the risk of doubling in serum creatinine and ESKD compared with placebo. Even with that precautionary approach, hospital admission for heart failure was higher in the atrasentan group, indicating the necessity for careful monitoring. ET-1 receptor A antagonists require a strategy to control fluid retention to enable safe use.
Currently, sparsentan, a dual-acting angiotensin and endothelin receptor antagonist that can inhibit both ET-A receptors and angiotensin II type 1 receptors of RAS, is being investigated in phase 3 clinical trials against focal segmental glomerulosclerosis (DUPLEX [A Randomized, Multicenter, Double-Blind, Parallel, Active-Control Study Of The Effects Of Sparsentan, A Dual Endothelin Receptor And Angiotensin Receptor Blocker, On Renal Outcomes In Patients With Primary Focal Segmental Glomerulosclerosis] trial) and immunoglobulin A nephritis (PROTECT [A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy] trial). When they become available, those results might provide insight into strategies for controlling fluid retention.
Pentoxifylline
Pentoxifylline is a nonspecific phosphodiesterase (PDE) inhibitor used clinically to treat peripheral vascular diseases. Inhibiting PDE activity increases cyclic adenosine-3,5-monophosphate levels and activates protein kinase A, leading to a reduction in proinflammatory cytokine production. Pentoxifylline modulates levels of TNF-α and other proinflammatory cytokines, including IL-1, IL-6, interferon γ, and other molecules such as ICAM1, vascular cell adhesion molecule 1 (VCAM1), and C-reactive protein [57]. Its anti-inflammatory effect could protect against DKD, and many clinical trials have shown that it has therapeutic effects in DKD. The PREDIAN (Pentoxifylline for Renoprotection in Diabetic Nephropathy) trial in patients with type 2 diabetes and CKD G3–4 taking standard RAS inhibitors reported that 2 years of pentoxifylline treatment significantly slowed the decline in eGFR and decreased albuminuria and urinary excretion of TNF-α [58]. Recently, the proinflammatory cytokines TNF-α and tumor necrosis factor-like weak inducer of apoptosis were reported to reduce renal Klotho expression mediated by NF-κB in mice and cell culture [59]. A recent post hoc analysis of the PREDIAN trial also showed a significant increase in serum and urinary Klotho concentrations [60]. This might also be related to the renal effects of pentoxifylline. A larger randomized clinical trial (gov Identifier: NCT03625648) is ongoing to examine the time to ESKD or death.
Apoptosis signal-regulating kinase-1 inhibitors
Apoptosis signal-regulating kinase (ASK)-1, a serine/threonine kinase, is an upstream signaling kinase of p38 MAPK and c-Jun N-terminal kinase (JNK). ASK-1 is normally bound and repressed by thioredoxin, an antioxidant protein. However, under oxidative stress, oxidized thioredoxin dissociates from ASK-1, activating it. The downstream p38 MAPK and JNK pathways induce apoptotic, inflammatory, and fibrotic signaling. The increased activation of p38 MAPK in DKD has been demonstrated in human renal biopsy analyses [61]. In an eNOS knockout mouse model of DKD, pharmacological ASK-1 inhibition halted the progressive decline in GFR and decreased albuminuria by reducing apoptosis and fibrosis within the glomerular and tubulointerstitial compartments and preserving podocyte density [61]. A phase 2 clinical trial of selonsertib, a selective ASK-1 inhibitor, found no difference in the primary outcome, change from baseline in eGFR at 48 weeks, between the selonsertib and placebo groups [62]. However, an unanticipated acute decline in eGFR was observed in the first 4 weeks in the 18-mg selonsertib group, which might have confounded the primary outcome. The acute effect of selonsertib on creatinine concentration is thought to be a result of inhibiting multidrug and toxin extrusion proteins 1 and 2 K. A post hoc analysis revealed that the rate of eGFR decline between 4 and 48 weeks was reduced by 71% for the 18-mg selonsertib group relative to the placebo group. The effects on the urine albumin-to-creatinine ratio were not confirmed. The MOSAIC (Study Evaluating the Efficacy and Safety of Selonsertib in Participants With Moderate to Advanced Diabetic Kidney Disease) phase 2b clinical trial is ongoing in type 2 diabetes patients with moderate to advanced DKD to evaluate whether selonsertib can slow the decline in kidney function.
C-C chemokine receptor 2 inhibitors
Chemokine C-C motif ligand 2 (CCL2), also known as monocyte chemoattractant protein-1, is a chemoattractant for monocytes, memory T cells, and natural killer cells that works by activating C-C chemokine receptor 2 (CCR2) and CCR4. CCL2 has been suggested as a potential marker for DKD [63]. Inhibiting CCL2 and CCR2 by various methods, such as neutralizing antibodies, receptor antagonists, inhibitors, DNA vaccines, mutant genes, and enantiomeric RNA oligonucleotides, decreases albuminuria, kidney damage, and inflammation in experimental kidney disease models, including DKD [64]. Some of the tested methods have progressed to clinical trials.
CCX140-B, a selective inhibitor of CCR2, was reported to reduce the urine albumin-to-creatinine ratio after 52 weeks of treatment in patients with type 2 diabetes on RAS inhibitors. In a phase 2 clinical trial, 5 mg of CCX140-B reduced the urine albumin-to-creatinine ratio by 16% compared with placebo without major side effects. The change in eGFR was not significant compared with the placebo group [65].
Inhibition of the CCL2/CCR2 pathway could be a promising therapeutic approach for DKD. However, more studies are needed to evaluate whether it can delay or halt the progression of DKD.
Janus kinase-1/2 inhibitors
The JAK-signal transducer and activator of transcription (STAT) pathway is involved in the transmission of signals such as cytokines and chemokines from extracellular ligands directly to the nucleus and induces a variety of cellular responses. The expression of multiple JAK-STAT family members is increased in the kidneys of DKD patients [66]. Moreover, preclinical experiments have shown that podocyte-specific JAK2 overexpression worsens renal injury [67], and a Stat3 knockdown model reduced albuminuria and renal inflammation in diabetic mice [68]. Based on that evidence for JAK-STAT activation in DKD, a phase 2 clinical trial with the JAK1/2 inhibitor baricitinib was conducted, and 24 weeks of baricitinib treatment significantly reduced albuminuria [69]. With baricitinib treatment, inflammatory biomarkers such as urinary C-X-C motif chemokine 10, urinary CCL2, plasma soluble tumor necrosis factor receptors 1 and 2, serum amyloid A, VCAM1, and ICAM1 decreased dose-dependently from baseline. The reduction in local inflammation might ameliorate DKD progression. However, no change in kidney function could be measured by serum creatinine, 24-hour urine creatinine clearance, or cystatin C-based eGFR. Furthermore, at 6 months, the high-dose baricitinib group had a statistically significant decrease in hemoglobin compared with the placebo group, as an important adverse event. Anemia might be attributed to a further decrease in the action of erythropoietin by JAK2 inhibition in those who already have low erythropoietin levels.
Other important regulators of JAK-STAT signaling are suppressors of cytokine signaling (SOCS). Enhancement of SOCS expression inhibits JAK-STAT signaling and could help prevent DKD progression [70].
Soluble guanylate cyclase stimulators and activators
Soluble guanylate cyclase (sGC) forms a heterodimer consisting of two subunits, α and β. Nitric oxide (NO) activates sGC by binding to the heme group present on the β-subunit, leading to increased cyclic guanosine monophosphate (cGMP). Increased cGMP promotes vasodilation and inhibits smooth muscle proliferation, leukocyte recruitment, platelet aggregation, and vascular remodeling [71]. NO/cGMP regulates renal blood flow, and increased cGMP dilates efferent arterioles, which seems to be beneficial for the prevention of glomerular damage and glomerulosclerosis. Increasing NO/sGC/cGMP signaling could inhibit kidney inflammation and fibrosis [72], potentially preventing or slowing the progression of DKD.
sGC stimulators and activators differ depending on the target. Heme in sGC is prone to oxidation under oxidative stress, which produces a heme-free form of sGC. sGC stimulators and sGC activators stimulate heme-containing sGC and heme-free sGC, respectively, in a NO-independent manner. Activating sGC signaling with cinaciguat, an sGC activator, markedly improved GFR, serum creatinine, mesangial expansion, and kidney fibrosis in streptozocin-induced diabetic eNOS knockout mice [73]. sGC activation by cinaciguat restored the glomerular cGMP content and reduced TGF-β1 expression and ERK1/2 phosphorylation, attenuating podocyte injury, proteinuria, glomerular cell proliferation, and apoptosis in a rat model of type-1 diabetes [74]. In a phase 2 trial, the sGC stimulator praliciguat did not significantly reduce albuminuria compared with placebo. On the other hand, praliciguat was associated with a modest reduction in blood pressure, hemoglobin A1c, and cholesterol, which could support further investigation of praliciguat in DKD [75].
Other medications in preclinical studies
Many other medications that target molecules related to DKD pathology are in development, and some of them have demonstrated promising effects in animal experiments.
For example, hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitors reverse the energy metabolism alterations that occur in the early stages of DKD in rats and mice and ameliorate the accumulation of glycolysis and tricarboxylic acid cycle metabolites in diabetic renal cortical tissue, which is thought to affect the progression of DKD [76]. In an obese type 2 diabetic mouse model, HIF-PH inhibitors improved insulin sensitivity, decreased albuminuria, and ameliorated glomerular epithelial and endothelial damage [77].
AGE-RAGE inhibitory modalities have therapeutic potential, but their clinical efficacy awaits confirmation. Similarly, approaches targeting epigenetics, such as histone demethylase inhibitors, DNA methyltransferase inhibitors, and pharmacological silencing of micro RNAs are under investigation, and some of them have shown renoprotective properties in animal experiments.
Conclusion
RAS inhibitors have been at the center of DKD treatment for a long time. Recent cardiovascular outcome trials of glucose-lowering agents, such as SGLT2 inhibitors and GLP-1 receptor agonists, have established the renoprotective roles of those agents. The safety and efficacy of nonsteroidal MRAs are being tested in large clinical trials. Uncovering the pathophysiology of DKD could allow the identification of novel therapeutic targets by which to prevent or slow the progression of DKD.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









