



Introduction
Mitochondria are double membrane-bound intracellular organelles essential for energy production in most eukaryotic cells [1]. Besides adenosine triphosphate (ATP) production, mitochondria also participate in various cellular processes such as redox homeostasis, intracellular calcium handling, cell differentiation, proliferation, and death [2]. A healthy and well-functioning mitochondrial population, achieved by so-called mitochondrial quality control, is therefore vital for cellular health [3]. Biogenesis, proteostasis, and mitophagy are critical components of mitochondrial quality control, and failure of mitochondrial quality control failure has been implicated in several human diseases [2,4].
Diabetes mellitus alters available energy substrates and results in excessive oxidative stress, followed by increased levels of inflammatory and profibrotic cytokines and cell death [5]. Abnormal mitochondria have been observed in renal biopsy specimens from diabetes patients [6ŌĆō9]. Urine metabolomic analysis has revealed universal depression of mitochondrial activity in patients with diabetic kidney disease (DKD) compared to diabetic patients without kidney disease [6]. Importantly, recent studies have demonstrated that mitochondrial dysfunction or abnormalities can mediate the initiation or aggravation of kidney injury in patients with diabetes [2,4]. In this review, we focus on the roles of mitochondrial quality control in DKD pathogenesis and discuss current research evidence and future directions.
Mitochondrial quality control
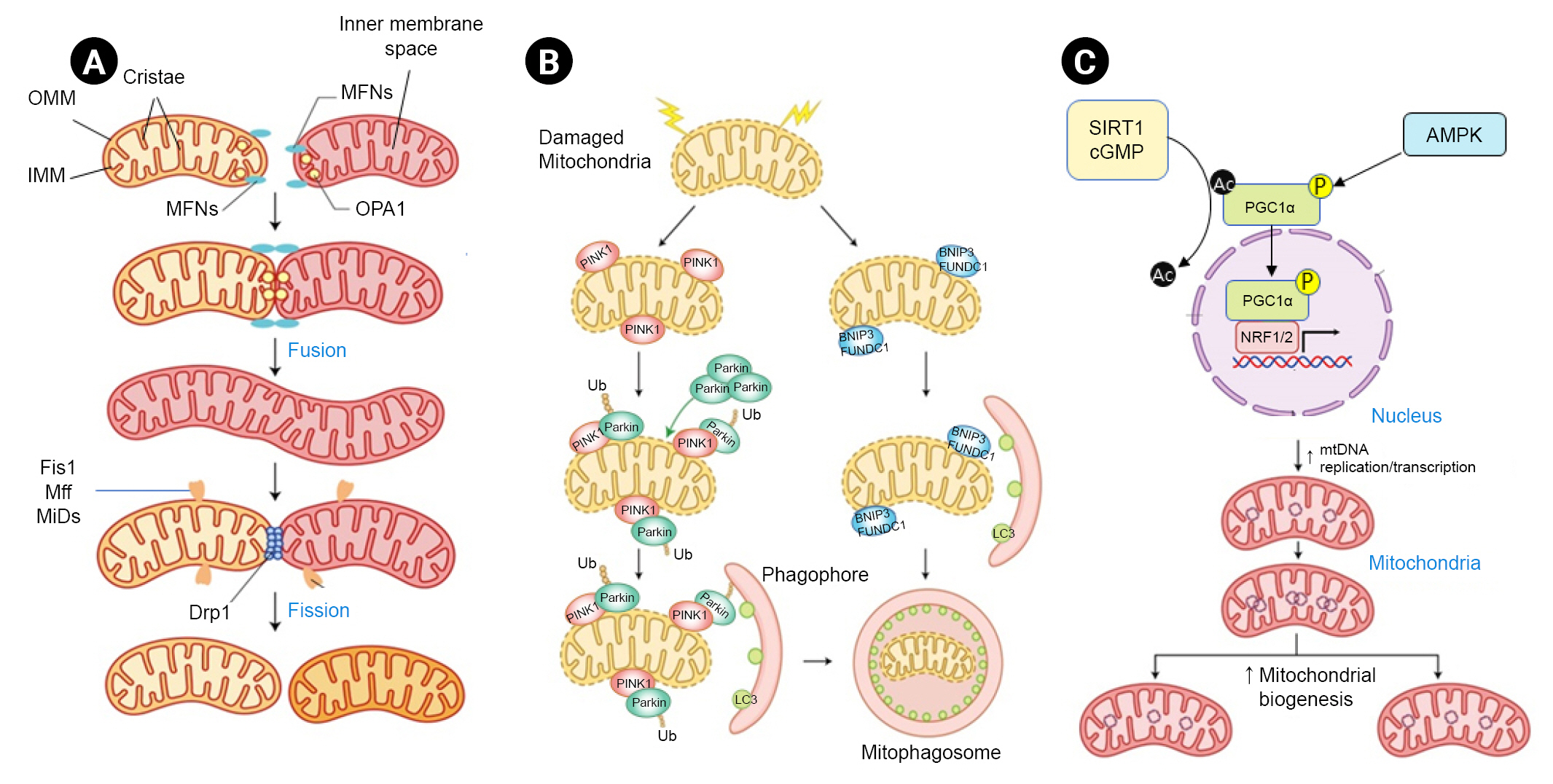
Mitochondrial quality control mechanisms comprise molecular and organelle quality control mechanisms that interact to maintain a healthy mitochondrial population [2,4]. Molecular-level mechanisms are composed of antioxidant defenses, protein quality control, and a DNA damage repair system (Fig. 1). Organelle-level mechanisms consist of mitochondrial fusion, fission, mitophagy, and mitochondrial biogenesis (Fig. 2).
Mitochondria are major sources of reactive oxygen species (ROS) due to oxidative phosphorylation, which is the metabolic process that results in ATP synthesis. In this context, mitochondria contain are enriched in antioxidants for redox homeostasis. When the balance between mitochondrial ROS production and the antioxidant system is disrupted, an oxidative stress situation develops with deleterious effects on organisms [10]. Mitochondria function as ROS amplifiers due to mitochondria-to-mitochondria and mitochondria-to-different ROS sources crosstalk with positive feedback [11]. Excessive ROS can lead to impairment of the electron transport chain (ETC) and low ATP production as a result of reduced cytochrome c oxidase activity [10]. In addition, defects in the ETC across the mitochondrial inner membrane can induce the loss of mitochondrial membrane potential and leakage of proapoptotic proteins into the cytosol [12]. Accumulation and decompartmentalization of mitochondrial ROS can result in oxidation of proteins, lipids, and DNA, leading to cellular dysfunction and disruption of crucial cellular signaling pathways [2,10].
The mitochondrial protein quality control system consists of two parts: the ubiquitin-proteosome system and chaperones [2,13]. Phosphatase and tensin homolog-induced putative kinase 1 (PINK1)-parkin signaling also contributes to mitochondrial protein quality control by inducing the formation of mitochondrial-derived vesicles or mitophagy [14]. Mitochondrial unfolded protein response (mtUPR) refers to the upregulation of mitochondrial proteases and chaperone gene transcription. This response is activated when the amount of unfolded or misfolded mitochondrial proteins overwhelms the capacity of the mitochondrial protein quality control system. Therefore, any defects in the mitochondrial protein quality control system are associated with failure of the mtUPR, and result in mitochondrial dysfunction and cell death [15].
Mitochondrial genome encodes 13 polypeptides essential for oxidative phosphorylation and ATP synthase; failure to repair damaged mitochondrial DNA can therefore result in insufficient ATP production and jeopardize cell survival [16]. Mitochondrial DNA has a 10- to 20-fold higher rate of mutagenesis than nuclear DNA because of its susceptibility to oxidative stress [17]. Absence of histone proteins, proximity to ROS production sites, and specific DNA replication through asymmetric patterns are possible explanations for the vulnerability of mitochondrial DNA to mutagenesis [2]. Mitochondrial DNA is repaired in the same manner as nuclear DNA [18].
Mitochondria maintain their abundance and functions through constant fusion and fission of the mitochondrial network [2]. Mitochondrial fusion allows the exchange of metabolites and DNA between mitochondria for their health, especially under metabolic and environmental stress [2,19]. In contrast, mitochondrial fission separates damaged and dysfunctional mitochondria from the mitochondrial network as a defense mechanism essential for mitochondrial regeneration, redistribution, and proliferation [2,20]. These dynamic mitochondrial morphological changes are controlled by expression of associated modulators and their posttranslational modification [2,3,19,20]. Mitochondrial fusion results in elongation of the mitochondrial network, which is associated with higher ATP production and maintenance of cell viability, whereas mitochondrial fission leads to short mitochondria, mitochondrial depolarization, and ROS overproduction with low ATP synthesis [3].
Mitophagy can selectively remove defective or surplus mitochondria from the mitochondrial network [21]. Several pathways are involved in labeling mitochondria and transferring them to autophagosomes [22]. The ubiquitin-mediated pathway is regulated by PINK1 and the E3-ubiquitin ligase parkin [2]. In the ubiquitin-independent receptor-mediated mitophagy pathway, autophagy receptor proteins can directly bridge with outer mitochondrial membrane proteins and microtubule-associated protein 1A/1B-light chain 3 (LC3B) [21,22].
Mitochondrial biogenesis is a complex process in which mitochondria increase in size and number to meet cellular energy demands [2]. Both mitochondrial DNA and nuclear DNA encode mitochondrial proteins. Therefore, elaborate coordination of cytoplasmic and mitochondrial protein synthesis is necessary [23]. Peroxisome proliferator-activated receptor-╬│ coactivator 1╬▒ (PGC1╬▒) plays an important role in directing this process [2,23]. PGC1╬▒ activity is controlled at both transcriptional and posttranslational levels [24].
Roles of mitochondrial quality control in diabetic kidney disease
After the heart, the kidneys, which function to remove waste products from blood, regulate fluid and electrolyte balance, and maintain blood pressure, contain the second largest number of mitochondria [3,23]. More than 20 types of specialized cells in mammalian kidney fulfill these important roles in the human body [25]. The nephron is the microscopic, functional unit of the kidney. Normally about 1 to 1.5 million nephrons are found in one kidney [25]. A nephron comprises a renal corpuscle (where urine formation begins by ultrafiltration) and a renal tubule responsible for absorption and excretion of various substances. In the renal corpuscle, fenestrated endothelium, glomerular basement membrane, and the foot process of podocytes comprise the filtration barrier and confer the nephron with charge and size-permselectivity. Renal tubules constitute approximately 90% of renal cortical cells and link the glomerulus to a collecting system. Defects in the filtration barrier and loss of nephrons result in proteinuria and a decreased glomerular filtration rate [26]. DKD is associated with a spectrum of characteristic morphologic changes of the glomeruli, tubules, interstitium, and vasculature [1].
Renal tubular cells
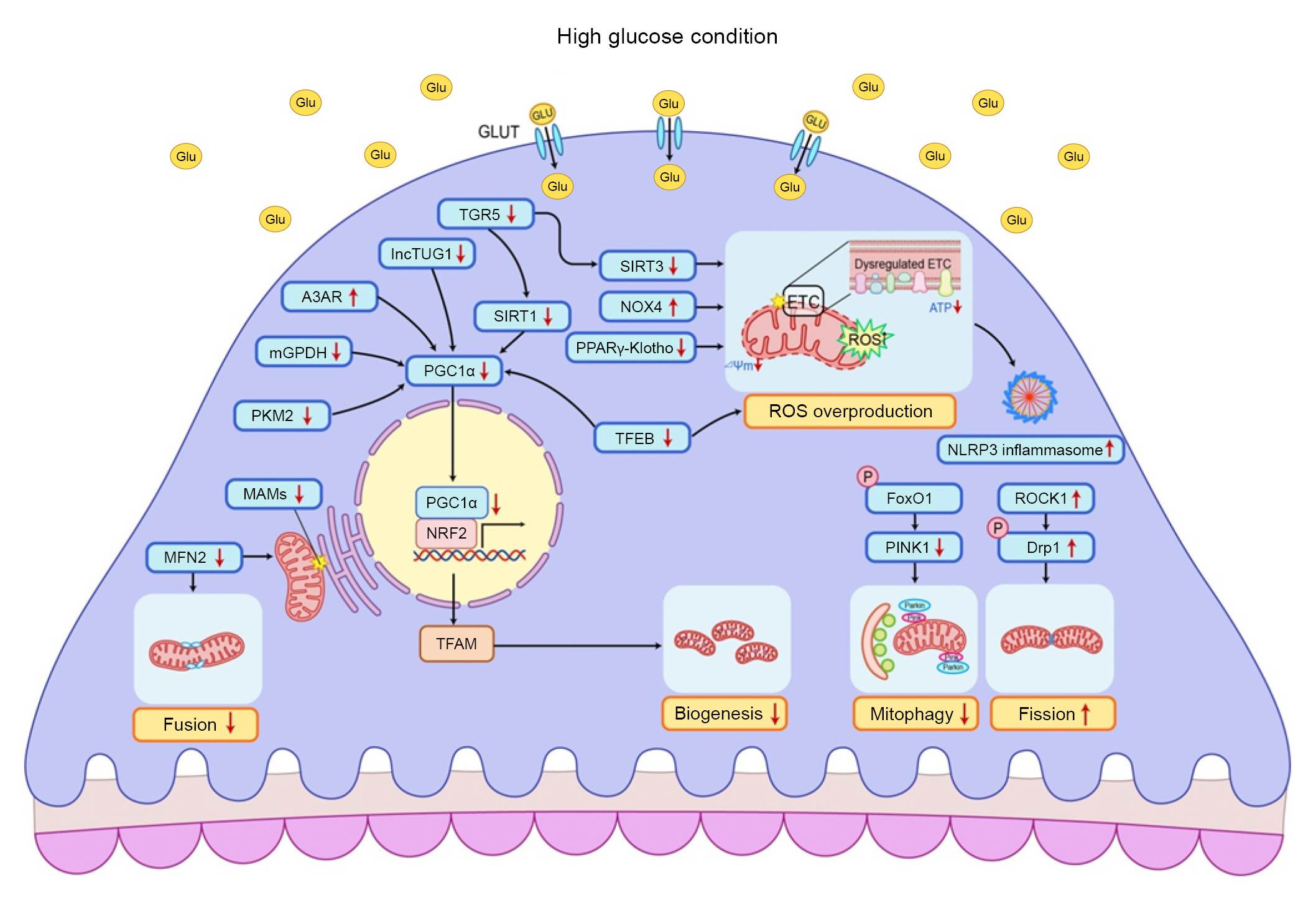
Renal tubular cells are rich in mitochondria, which are required to generate ATP and power tasks such as active reabsorption of sodium, glucose, and other metabolites from urine [5]. Diabetic milieu can promote renal tubular cells to utilize more ATP than usual to increase active reuptake of urinary metabolites, particularly glucose reabsorption via the sodium-glucose cotransporter 2 (SGLT2). Due to the increased intracellular energy requirement, transport of fuel substrates such as free fatty acids to renal tubular cells is significantly increased in patients with diabetes [27]. Thus, any cellular event that decreases the fuel supply or increases oxygen demand (i.e., supply-demand imbalance) is an important pathophysiological contributor to diabetic tubulopathy in DKD [28]. Interestingly, the properties of mitochondria in proximal and distal tubules differ [29]. Studies using multiphoton live imaging of rat kidneys have shown that mitochondria in proximal tubules are more numerous but have lower membrane potentials and greater alterations in ROS generation upon ETC inhibition than those in distal tubules [29]. These properties may account for the clinical fragility of proximal tubules when mitochondrial homeostasis is disrupted. Fig. 3 shows how mitochondrial quality control in renal tubule cells is altered under high glucose conditions.
Excessive mitochondrial ROS can be induced by hyperglycemic stimuli, and these can contribute to diabetic tubulopathy [30]. An experimental study using rat renal proximal tubular cells demonstrated that high glucose media can induce the overproduction of mitochondrial superoxide, change the mitochondrial membrane potential, and decrease ATP generation with complex III dysfunction, culminating in tubular cell death [30]. It has been suggested that renal mitochondrial oxidative stress is exacerbated by reduced sirtuin 3 activity in Zucker diabetic fat rats (ZDFRs) [31]. Conversely, a CD38 (cluster of differentiation 38) inhibitor was shown to restore the intracellular NAD+/NADH ratio and sirtuin 3-mediated mitochondrial antioxidative enzyme activity in the kidneys of ZDFRs [32]. Sirtuin 3 functions as an antioxidant in mitochondria by activating isocitrate dehydrogenase 2 and superoxide dismutase 2 [32]. A recent study reported that hypoxia-inducible factor 1 (HIF1) is involved in mitochondrial redox homeostasis in diabetes [33]. Zheng et al. [33] showed that HIF1 activity was suppressed and that blood ROS levels increased in response to hypoxic exposure in patients with type 1 diabetes. They also demonstrated that hyperglycemia could diminish the HIF1 response in renal tubular cells under hypoxia and in kidneys from diabetic animals. Low HIF1 levels are associated with mitochondrial oxidative stress through increased mitochondrial respiration. In addition, it has been suggested that HIF1 can improve mitochondrial homeostasis via heme oxygenase 1 in renal tubules in diabetic environments [34].
Mitochondrial dynamics are profoundly altered in DKD. An increase in mitochondrial fragmentation has been shown to be an early phenomenon in renal tubules of experimental diabetes models [35]. Mitofusin (MFN) 1, a mitochondrial fusion-related protein, has been shown to be downregulated whereas dynamin-related protein 1 (Drp1), a pro-fission protein, has been shown to be upregulated in human renal proximal cells (HKC8) under high glucose conditions as well as in the kidneys of diabetic mice [36]. Several mechanisms have been suggested to mediate mitochondrial fragmentation in diabetic tubular injury [37ŌĆō39]. Liu et al. [37] demonstrated that hyperglycemia can inhibit AMP-activated protein kinase (AMPK) phosphorylation and induce an increase in SP1 (specificity protein 1) followed by PGAM5 (phosphoglycerate mutase family member 5) upregulation, resulting in Drp1-dependent mitochondrial fission in diabetic renal tubular injury. Zhang et al. [38] demonstrated that stromal cell-derived factor-1╬▒ (SDF-1╬▒)/CXC chemokine receptor 4 (CXCR4)/signal transducer and activator of transcription 3 (STAT3) signaling is associated with mitochondrial dysfunction in diabetic tubulopathy. In this regard, sitagliptin, a DPP4 (dipeptidyl peptidase 4) inhibitor, can improve mitochondrial dynamics in diabetic tubular injury by restoring SDF-1╬▒/CXCR4/STAT3 signaling and activating OPA1 (optic atrophy 1). Increased mitochondrial fragmentation and changes in MFN1 and mitochondrial fission 1 protein (Fis1) expression have also been observed in proximal tubular cells of a DKD patient [39]. It has been suggested that p66Shc (Src homologous and collagen) can mediate hyperglycemia-induced mitochondrial fragmentation and apoptosis signaling [39].
Mitophagy plays a protective role against cell damage. Inappropriate mitophagy is associated with hyperglycemia-induced cytotoxicity [1,21] while reduction of mitophagy is associated with accumulation of intracellular mitochondrial ROS. Overexpression of myo-inositol oxygenase (MIOX), an enzyme that inhibits PINK1-parkin-mediated mitophagy, increased mitochondrial ROS production in high glucose-treated renal tubular cells [7]. In contrast, mitochondrial-targeting antioxidant mitoquinone mesylate was shown to restore mitophagy activity and reduce tubular cell death through restoration of nuclear factor erythroid 2-related factor 2 (NRF2) and PINK1 activity in an experimental DKD model [40]. Studies on the mechanism of mitophagy regulation in diabetic tubulopathy are ongoing. Thioredoxin interacting protein has been reported to inhibit autophagy/mitophagy flux via mammalian target of rapamycin signaling in renal tubular cells under high glucose conditions [41]. Overexpression of optineurin, a coordinator protein linking damaged mitochondria and autophagy, was shown to promote mitophagy and relieve cellular senescence in renal tubular cells of diabetic mice [42]. It has been suggested that an increase in the long noncoding RNA (lncRNA) NEAT1 (nuclear paraspeckle assembly transcript 1) can inhibit mitophagy via the miR150-5p-Drp1 axis in high glucose-exposed HK2 human proximal tubular cells [43]. Recent studies have shown that upregulation of TIPE1 (tumor necrosis factor alpha-induced protein 8-like 1) in tubular epithelial cells can interfere with PHB2 (prohibitin 2)-mediated mitophagy and exacerbate diabetic tubulopathy [44]. Melatonin, a pineal hormone involved in regulation of circadian rhythms, can ameliorate diabetic tubulopathy via the AMPK-PINK1-mitophagy pathway in HK2 cells and streptozotocin (STZ)-induced diabetic mice [45].
Mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) play a critical role in mitochondrial quality control during ER stress [46]. MAMs are a region of interaction between the ER and mitochondria. They have diverse functions such as nutrient and hormone signaling, regulation of calcium homeostasis, autophagy, and apoptosis [46]. Li et al. [46] demonstrated that phosphofurin acidic cluster sorting protein 2 (PACS-2) expression is decreased in renal tubules of patients with DKD and that PACS-2 overexpression in HK2 cells can restore MAM integrity and promote the formation of mitophagosomes.
Analytical studies of human and animal samples have demonstrated inefficient mitochondrial bioenergetics and reduced functional mitochondrial mass in DKD [6,47,48]. PGC1╬▒, a key regulator of mitochondrial biogenesis, is highly expressed in renal proximal tubules where mitochondria are abundant. Notably, failure of PGC1╬▒ to be upregulated under hyperglycemic circumstances can aggravate diabetic tubulopathy [49]. Therefore, enhancing PGC1╬▒ activity could be a potential therapeutic strategy for treating DKD. Reduction of PGC1╬▒ expression is accompanied by aberrant mitochondrial dynamics, excessive ROS production, and tubular cell death, whereas administration of the pharmacological PGC1╬▒ activators 5-aminoimidazole-4-carboxamide-1-╬▓-D-ribofuranoside (AICAR) or metformin can restore mitochondrial homeostasis and reduce apoptosis. Beneficial effects of AICAR and metformin have been confirmed in STZ-induced diabetic mice; AICAR and metformin increase renal PGC1╬▒ expression, improve mitochondrial fragmentation and ROS production, and improve hyperglycemia-related tubular damage and renal fibrosis [47]. In addition, chloroquine and amodiaquine (traditionally used to treat malaria) can effectively abrogate downregulation of AMPK and PGC1╬▒ phosphorylation due to hyperglycemia, restore mitochondrial homeostasis, and alleviate albuminuria and renal histopathological changes in the kidneys of diabetic mice [48,50].
Interestingly, the PGC1╬▒-induced increase in mitochondrial biogenesis affects mitochondrial ROS generation, dynamics, and autophagic elimination of mitochondria, suggesting complex, synergistic interactions among various mechanisms for adequate quality control of mitochondria in diabetic tubulopathy.
Podocytes
Podocyte injury is a critical event in the development of DKD [51]. As podocytes are terminally differentiated cells, their loss is irreversible, which can lead to functional decline of the glomerular filtration barrier [52]. Podocytes are rich in mitochondria because they require high levels of energy to maintain the foot processes [52]. As with renal tubular cells, podocytes under high glucose conditions show defects in mitochondrial quality control such as mitochondrial ROS overproduction, abnormal dynamics, inappropriate mitophagy, and failure of biogenesis. Fig. 4 shows how mitochondrial quality control is altered in podocytes under high glucose conditions.
Hyperglycemia can induce excessive ROS production from mitochondria and contributes to podocyte apoptosis coinciding with albuminuria [53]. Peroxisome proliferator-activated receptor (PPAR) signaling is downregulated in podocytes under high glucose conditions. Use of PPAR activators such as thiazolidinediones is one strategy for recovery of insulin resistance in diabetic patients. Pharmacologic augmentation of the PPAR╬│-Klotho axis can help attenuate diabetic podocytopathy via suppression of mitochondrial ROS overproduction and activation of NRF2 signaling [54ŌĆō56]. NADPH oxidase 4 (NOX4) is the principle non-mitochondrial ROS source in diabetic kidneys. You et al. [57] demonstrated that hydrogen peroxide produced by NOX4 can inhibit mitochondrial fumarate hydratase followed by upregulation of HIF1╬▒ signaling, which is associated with inflammatory and profibrotic cytokines. Interestingly, NOX4 also appears to be present in the mitochondria of podocytes [58]. Overproduction of ROS from mitochondrial and non-mitochondrial sources can oxidize cardiolipin, a mitochondrial membrane-specific phospholipid, which is an initiator for the NLR family pyrin domain containing 3 (NLRP3) inflammasome [59]. Activation of the NLRP3 inflammasome can lead to caspase 1-dependent release of proinflammatory cytokines. Persistent inflammation induced by the NLRP3 inflammasome pathway plays an important role in the pathophysiology of diabetic podocytopathy [60].
Unopposed mitochondrial fission in podocytes is a pathognomonic feature of diabetic podocytopathy and is present before the clinical manifestations of DKD [61,62]. When the mitochondrial fission regulator Drp1 is deleted in podocytes, albuminuria is decreased and major pathologic features of DKD are improved in mice [62]. Wang et al. [61] found that Rho-associated coiled-coil containing protein kinase 1 (ROCK1) contributes to Drp1 recruitment to mitochondrial membrane by phosphorylation of Drp1 and aggravates mitochondrial fission and ROS production in podocytes under high glucose conditions. Podocyte-specific deletion of ROCK1 can inhibit apoptosis and mitochondrial ROS production [61]. MFN2, a fusion-related protein, is located at ER membranes and regulates the dynamics of MAMs [8]. Cao et al. [63] demonstrated that MFN2 expression was low in the podocytes of DKD patients and that MFN2 overexpression attenuated the high glucose effect in an animal model. They also confirmed that MFN2-protein kinase RNA-like ER kinase (PERK)-regulated MAMs and had antiapoptotic effects in podocytes under hyperglycemic conditions [8].
A few studies have investigated the role of mitophagy in diabetic podocytopathy. Mitophagy is an important defense mechanism during cellular stress, and inappropriate mitophagy can exacerbate podocyte injury [64,65]. Forkhead transcription factor O1 (FoxO1) is inactivated by the PI3K/Akt (phosphatidylinositol-3-kinase/Akt) pathway under high glucose conditions. FoxO1 upregulation can increase PINK1/parkin-dependent mitophagy to restore podocyte damage in STZ-induced diabetic type 1 diabetic mice [64]. Progranulin is an autocrine growth hormone involved in development, inflammation, cell proliferation, and protein homeostasis [65]. Recombinant human progranulin treatment was shown to facilitate mitophagy and mitochondrial biogenesis in the podocytes of diabetic mice through progranulin-sirtuin1-PGC1╬▒/FoxO1 signaling [65].
Mitochondrial biogenesis is suppressed in podocytes under high glucose conditions [9,66]. Reduced expression of PGC1╬▒ and mitochondrial dysfunction have been observed in the podocytes of DKD animal models [9,66]. Analysis of kidney biopsies from patients with DKD revealed a reduction in PGC1╬▒ expression in micro-dissected glomeruli [67]. As a positive control for PGC1╬▒, podocyte-specific knockdown of sirtuin1 can increase susceptibility to diabetic nephropathy in a murine model [68]. In contrast, podocyte-specific sirtuin1 overexpression can ameliorate podocyte loss and DKD progression [69]. lncRNA taurine upregulated gene 1 (TUG1) is a recently discovered PGC1╬▒ regulator. Podocyte-specific overexpression of TUG1 in diabetic mice restored PGC1╬▒ expression, leading to improved mitochondrial ATP production along with amelioration of diabetic glomerulopathy [9]. Proteomic analysis using glomeruli from individuals with or without DKD has revealed that levels of pyruvate kinase M2 (PKM2), a rate-limiting glycolytic enzyme, are elevated in diabetes patients without DKD [70]. Podocyte-specific PKM2 knockout mice with diabetes showed aggravated albuminuria with more severe pathologic changes in the glomeruli than wild-type diabetic mice. Conversely, treatment with a PKM2 activator was shown to induce mitochondrial biogenesis and reverse glomerular pathology in part by increasing glycolytic flux and PGC1╬▒ transcription [70]. Factors that affect mitochondrial biogenesis in diabetic podocytopathy by acting directly or indirectly on PGC1╬▒ such as TGR5 (G protein-coupled bile acid receptor), A3 adenosine receptor, transcription factor EB, and mitochondrial glycerol 3-phosphate dehydrogenase are continuously being discovered [71ŌĆō74]. NRF2 is a downstream transcription factor of PGC1╬▒. It is responsible for gene expression in the mitochondrial redox system and the induction of TFAM (mitochondrial transcription factor A) [2]. Wang et al. [75] showed that NRF2 activation attenuated high glucose-induced injury in mouse podocytes and that downregulation of NRF2 promoted more severe injury and increased ROS production in podocytes exposed to high glucose levels.
Evidence published to date indicates that mitochondrial quality control plays an important role in diabetic podocytopathy. There are several unanswered questions in this area. For example, the precise roles and interactions of each process involved in mitochondrial quality control and the detailed role of MAMs during the development and progression of diabetic podocytopathy require further exploration.
Endothelial cells
In DKD, glomerular endothelial cell injury is an early process that occurs before the onset of albuminuria. This event contributes to DKD by paracrine mediator release, leading to subsequent kidney injury [76]. Although the mechanism of glomerular endothelial cell-podocyte crosstalk regulation in DKD is obscure, a previous study showed that podocyte detachment and a decrease in endothelial cell fenestration play an important role in kidney injury in type 2 diabetes [77]. In addition, conditioned medium derived from endothelial nitric oxide synthase-deficient glomerular endothelial cells induced podocyte injury under high glucose conditions, suggesting the importance of communication between endothelial cells and podocytes in diabetes [78]. Qi et al. [79] investigated the role of glomerular endothelial mitochondrial dysfunction in DKD. They found that mitochondrial dysfunction and oxidized mitochondrial DNA in glomerular endothelial cells were related to high glucose-induced podocytopathy after comparing transcriptome profiles between DKD-resistant and susceptible mice strains [79]. They demonstrated that diabetes-induced endothelin-1 (Edn1)/Edn1 receptor type A (Ednra) signaling facilitated mitochondrial stress and injury in endothelial cells [79]. They also observed that mitochondrial DNA damage resulting from mitochondrial ROS was associated with glomerular endothelial Ednra expression and rapid DKD progression. Notably, an Ednra antagonist inhibited endothelial mitochondrial oxidative damage and reduced albuminuria and podocyte depletion in DKD-susceptible mice [79].
Hyperglycemia can lead to mitochondrial fission in glomerular endothelial cells coupled with mitochondrial ROS production and cell death [61]. ROCK1 contributes to Drp1 activation by phosphorylating Drp1 at serine 600 and triggers mitochondrial fragmentation, as it does in podocytes under high glucose conditions [61].
Although the content of mitochondria in endothelial cells is low, mitochondria play an important role in endothelial cell signaling in response to external stimuli. Therefore, studies on mitochondrial quality control of endothelial cells in diabetic nephropathy are necessary for new drug development.
Immune cells
Intrarenal inflammation is one of the most important contributing factors to the initiation and progression of DKD. Given that immune cell infiltration is frequently observed in biopsy-proven DKD, it is a major pathologic criterion used in pathologic classification of DKD [80]. Research studies have consistently demonstrated that the severity of interstitial inflammation is significantly associated with renal outcomes in patients with DKD [81ŌĆō83]. Notably, kidney transcriptomic profiles of human diabetic kidneys have revealed decreased expression levels of inflammation-associated genes in early diabetic kidneys compared to healthy controls, whereas expression levels of these genes are significantly increased in advanced diabetic kidneys, suggesting that the roles of inflammation in early and late DKD are different [84]. Deconvolution of RNA-sequencing data revealed significantly increased infiltration of virtually all immune cell types including macrophages, monocytes, B and T cells, and plasma cells, in advanced DKD compared to early DKD and control samples [84]. The significant increase in proinflammatory stimuli in patients with advanced DKD suggests that intrarenal inflammation plays a pivotal role in the progression of DKD. Consistent with these data, several studies have demonstrated that intrarenal macrophage infiltration is associated with albuminuria, the severity of histologic damage, and adverse renal outcomes [83,85].
Hyperglycemia can significantly affect mitochondrial homeostasis in immune cells and induce phenotypic changes. Several studies have shown that hyperglycemic stimuli can provoke mitochondrial dysfunction in circulating macrophages, which in turn can increase the proportion of proinflammatory M1 macrophages [86]. Hyperglycemia can induce the proinflammatory polarization of T cell compartments by increasing Th17 and Th1 subsets and decreasing regulatory T subsets [87]. Hyperglycemia can also induce mitochondrial inner-membrane hyperpolarization and increase the production of intracellular ROS and activation-induced interferon-╬│ [88]. Several studies have shown that B cells can also acquire proinflammatory phenotypes upon hyperglycemic stimulation. B cells have also been suggested to be involved in the pathogenesis of DKD, mainly by producing antibodies that can lead to the formation and deposition of immune complexes in the kidney [89]. Nevertheless, the role of immune cell mitochondria in DKD requires further investigation.
Conclusions and future perspectives
Defects in mitochondrial quality control and mitochondrial dysfunction are common characteristics of damaged kidney cells under hyperglycemic conditions [4,90]. Studies using diabetic rats have shown that abnormal changes in mitochondria precede the onset of albuminuria and histopathology [35]. Thus, disruption of mitochondrial homeostasis has been postulated to be a primary initiator of DKD and a potential target for developing new drugs. However, most studies that have investigated mitochondrial homeostasis-targeting drugs for DKD are in preclinical trials, with the exception of bardoxolone methyl [91]. Bardoxolone methyl, an activator of the NRF2 pathway, can inhibit mitochondrial ROS generation and nuclear factor kappa B signaling and is currently being investigated in a phase 3 clinical trial for DKD patients (NCT03550443).
Interestingly, although newly introduced antidiabetic drugs with reno-protective effects were not initially developed to target mitochondria, subsequent studies have shown that they improve mitochondrial health and dynamics [36,37,92]. In vivo and in vitro experiments have revealed that SGLT2 inhibitors can reduce mitochondrial fission and facilitate mitophagy in DKD [36,37]. Similarly, analysis of urine metabolites in patients with type 2 diabetes mellitus treated with atrasentan (an endothelin A receptor antagonist) suggested that this drug can prevent renal mitochondrial dysfunction under hyperglycemic conditions [92]. Therefore, the ability of a drug to affect mitochondrial dynamics and function may be used to determine its potential therapeutic effectiveness in DKD.
Due to the increasing prevalence of diabetes worldwide and the adverse effects of DKD on mortality, it is critical to gain a comprehensive understanding of the pathogenesis of DKD. Although many studies have investigated mitochondrial quality control, much remains to be clarified. Mitochondrial quality control deserves further investigation as a potential novel therapeutic target for DKD.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









